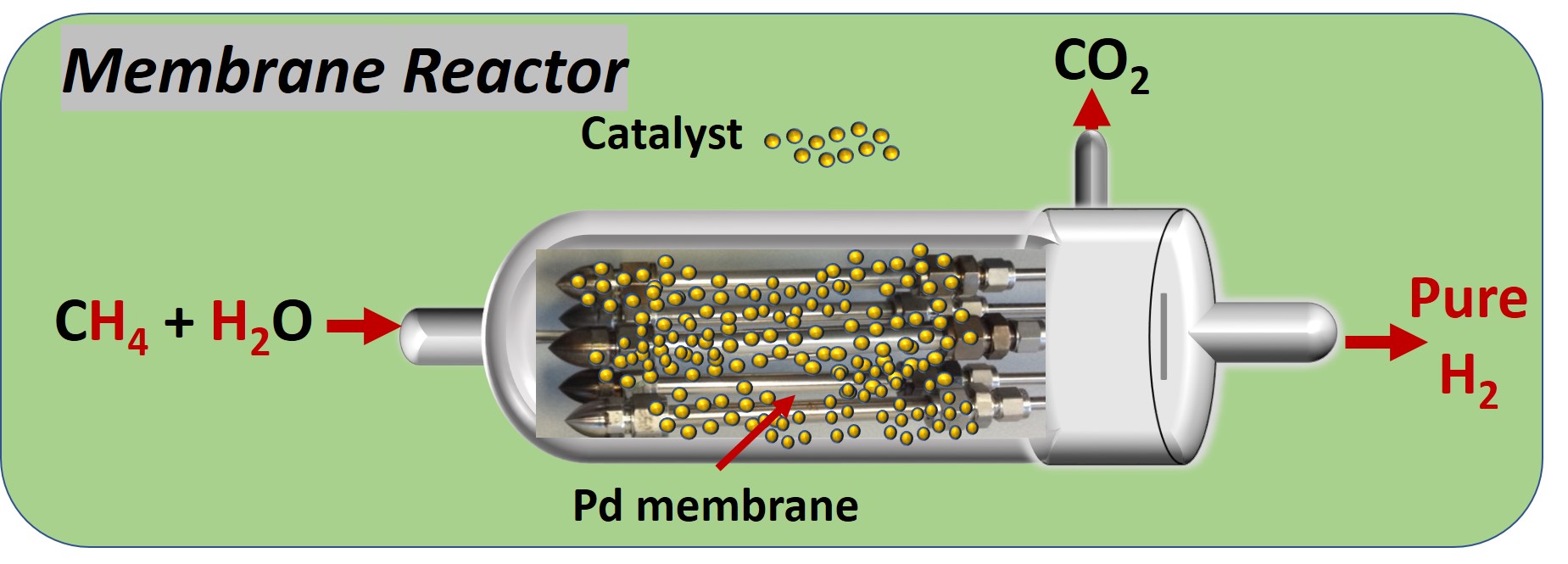
Among the main sustainable development goals of the European Union (EU) is a clean planet for all – the long-term vision for a prosperous, modern, competitive and climate neutral economy by 2050. The EU continues the path to a low-carbon, climate-neutral, resource-efficient and biodiverse economy in full compliance with the United Nations 2030 agenda. The adoption of process intensification methodologies that use the integration of reaction and separation in single, multifunctional units, will improve energy efficiency and competitiveness. One of these technologies is the membrane reactor (MR), where reaction and separation through membranes are integrated to achieve higher yields at lower CAPEX and OPEX.
Using selective membranes one product is remove from the reaction zone, shifting the reaction to the products, which increases the yield while also obtaining a pure product.
Among the EU projects working on MRs, FERRET, FluidCELL, BIONICO apply the MR concept for hydrogen production in a micro-CHP (combined heat and power) systems using both natural gas and biofuels (biogas and bioethanol). Hydrogen obtained from the reforming Palladium MR is used in a PEM fuel cell to produce electricity. Pd-MR were also used for the product hydrogen from syngas (DEMCAMER, CACHET II) and propene dehydrogenation (CARENA). Similarly, water gas selective membranes for CO2 hydrogenation to produce methanol using zeolite-MR (CARENA), or carbon-MR DME (C2FUEL). Oxygen membranes are used for the controlled delivered of oxygen for direct conversion of methane into ethylene (MEMERE). Supported ionic liquids membranes are used in ROMEO to improve the hydroformylation reaction. In this talk we will discuss the latest results of these EU projects on MRs.
An energy career medium is required for the world-wide utilization of renewable energy. Ammonia is one of the promising energy career medium, and carbon-free hydrogen is easily produced via ammonia decomposition. Ammonia as well as produced hydrogen will be used as energy for power generation; however, methane synthesized by reacting the produced hydrogen with CO2 will be easily used in the present infrastructure. The most serious demerit of methanation is exothermic reaction; however, combined reaction system of NH3 decomposition and CO2 methanation, heat from exothermic methanation will be used effectively for endothermic ammonia reaction,
In this study, combined reactions of NH3 decomposition and CO2 methanation were conducted at 623 K in a hydrogen-permeable palladium membrane reactor, where heat transfer as well mass transfer of hydrogen were possible. Even if commercial Ru catalyst was used for both NH3 decomposition and CO2 methanation, the levels of NH3conversion and CO2methanation were increased by membrane reactor effect, namely effective hydrogen permeation from NH3 decomposition side to CO2 methanation side.
In order to improve the performance of combined reaction system, novel catalysts were prepared for NH3 decomposition and CO2 methanation at relatively low reaction temperature of 623 K. Among the catalysts developed, Ru/BaO/Al2O3 and Ru/ZrO2 gave high catalytic activity for NH3 decomposition and CO2 methanation, respectively. As a result of the improvement of both hydrogen production rate in NH3 decomposition and hydrogen consumption rate in the CO2 methanation, the increased difference in hydrogen partial pressure led to acceleration of hydrogen permeation. Furthermore, the reaction fields of ammonia decomposition and CO2 methanation were interchanged on the inside and the outside of the hydrogen permeable membrane in consideration of their catalytic activity. It was experimentally demonstrated that membrane reactor effect was improved by development of catalysts tin the combined reaction system.
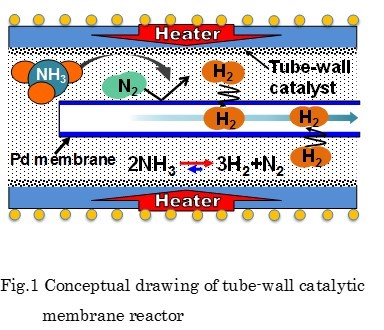
As an efficient and convenient mean of transporting and storing hydrogen, it is proposed that hydrogen is converted into chemical compounds such as ammonia, which is called hydrogen carrier. Among the carrier candidates, ammonia has some advantages like possessing a comparatively large hydrogen storage capacity (17.7wt%), being liquefied easily at 20 °C and 0.8MPa, and emitting no CO2. However, to produce hydrogen by ammonia decomposition a higher temperature, usually over 600 °C, is needed because of endothermic and slow reaction.
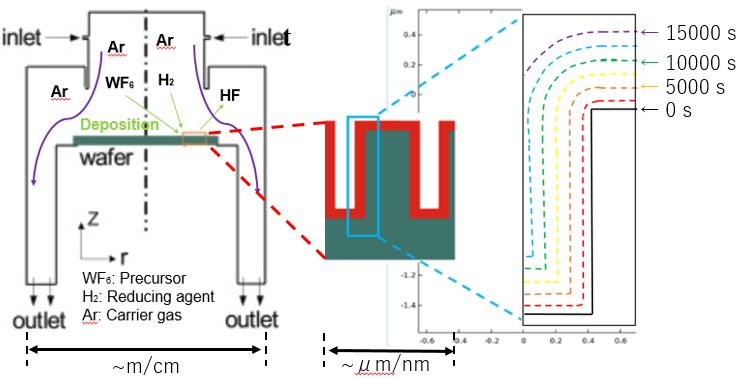
In this study, a new type of membrane reactor that consists of a tube-wall reactor and a palladium membrane tube as shown in Fig. 1 is proposed for hydrogen production via ammonia decomposition. This reactor, in which uniform and direct heat supply can be realized compared with a conventional packed bed reactor, will allow ammonia to decompose efficiently.
A tube-wall reactor was made from aluminum tube with 8mm in inner diameter, 1mm thick and 100mm long. The inside wall was anodized to 0.1mm-thick alumina layer for the support of Ru catalyst, where the evaporation-to-dryness method using RuCl3 aqueous solution was applied for the Ru loading. Ether a rolled Pd77Ag33 tube (3.2mm in outer diameter, 0.2mm thick and 100mm long) or a composite Pd/Al2O3 tube (0.002mm thick and 90mm long), prepared by chemical vapor deposition was inserted at center of the tube-wall reactor. Ammonia decomposition was carried out in the range of 325 - 425 °C at 0.1 MPa to compare the differences between 1) the packed-type and the tube wall-type, and 2) the tube-wall reactors with and without hydrogen separation by Pd membrane. When performances between the tube-wall reactors with and without hydrogen separation by the 0.2mm-thick rolled Pd alloy membrane were compared, an increment of conversion was clearly observed.
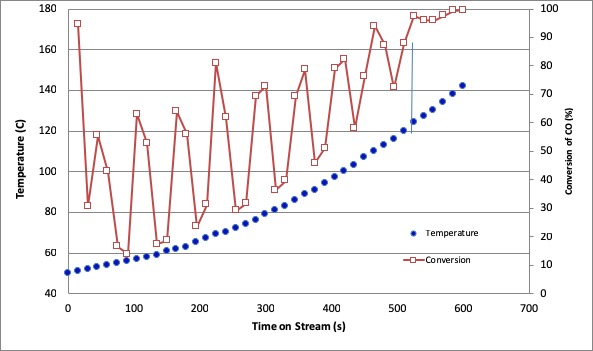
CO oxidation process in the catalytic converter hasn't showed best performance particularly when it is cold start-up, since the catalyst inside the converter is not active during this period. The purpose of this experiment was to develop the forced unsteady state operation procedure of CO oxidation using Pt/γ-Al2O3 with 0.05%-w and space velocity of 0.406 mmol s-1gram-1catalyst. The catalytic converter was gradually ramped-up, while introducing the feed gas containing CO in the air. The feed gas was modulated following a square wave model with switching time variation at 3, 6, 15, and 30 s and various operation modes. To gain the intrinsic reaction rate, the external mass transfer criterion was determined. Ramping-up the temperature from 50 until 150 °C increased the CO conversion with different profiles between steady state and dynamic flow rate. The dynamic system with modulated CO feed flow gave lower light-off temperature and higher average CO conversion than the steady state system which gave light off temperature 115 °C and average CO conversion of 48.86 %. The switching time of 3 s gave highest average CO conversion during ramping-up, which was 79.35 %. Meanwhile the dynamic operation system with modulated compressed feed flow gave higher light off temperature and lower average CO conversion than steady state system.
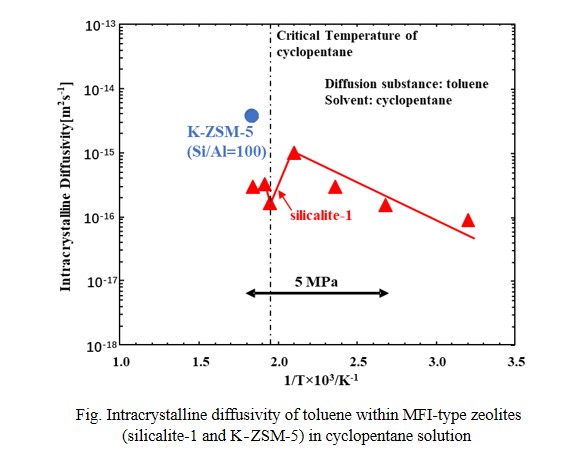
In this study, intracrystalline diffusivity of toluene within MFI-type zeolites (silicalite-1 and K-ZSM-5 (Si/Al=100)), possessing micropore which size is close to molecular size of toluene, in sub-, and super-critical fluid of cyclopentane was investigated using the constant volumetric method. This study clarified that concentration of toluene in sub- and super-critical fluid of cyclopentane (critical point: 511K, 4.5MPa) can be measured using Raman spectroscopy. Using change in toluene concentration with time due to adsorption of toluene onto the zeolite and the intracrystalline diffusivity was calculated using theoretical equation. Logarithm of intracrystalline diffusivities of toluene within silicalite-1 (MFI-type zeolite without Al in its framework) measured under 5 MPa was correlated with a single line against inverse number of temperatures. In contrast, above 473 K, the intracrystalline diffusivity of toluene within silicalite-1 was decreased and took minimum value around critical temperature. Formation of clusters between solvent molecules and diffusion substance near the critical point of solvent was considered to lead to the increase of diffusion resistance at pore mouth of silicalite-1. In addition, the intracrystalline diffusivity of toluene within K-ZSM-5 in supercritical fluid of cyclopentane at 543K was also measured. The intracrystalline diffusivity showed much higher value than that within silicalite-1. By increasing toluene concentration at outer surface of the zeolite, formation of the cluster may be suppressed and decreased diffusion resistance at pore mouth, which leading to increase the diffusivity.
Metal oxides such as HfO2, ZrO2 and Y2O3 have optical, mechanical and chemical properties which raise interest for a variety of applications in fields such as sensors, optoelectronics, medical devices.... Atomic Layer Deposition (ALD) provides excellent uniformity, conformality in complex topographical structures, as well as good film purity. However, ALD is heavily dependant on the precursor choice, which may both determine film performances as well as the process Cost-of-Ownership with considerations around thermal stability, volatility, physical state. In this presentation, we will introduce recent developments on Group IV metal oxide precursors (high-temperature HfO2, ZrO2 ALD) as well as liquid lanthanide compounds (Y2O3, Sc2O3, Ln2O3) and illustrate films performances for applications such as hydrophobic coatings, anti-corrosion coatings and optical coatings.

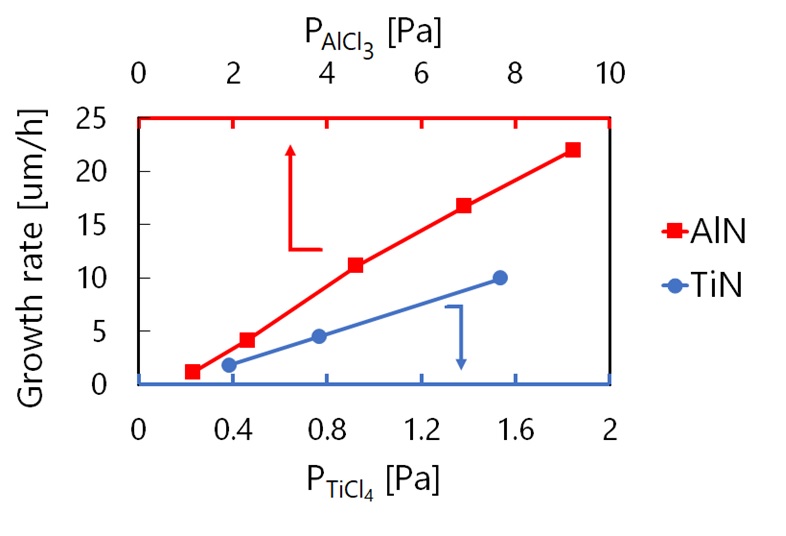
Aluminum-rich cubic titanium aluminum nitride (Ti1-xAlxN, x>0.9) is an emerging material for cutting tool coating due to high hardness, thermal resistance, and chemical stability. Cubic phase is, however, metastable at x>0.67 (hexagonal phase most stable), and physical vapor deposition (PVD) consistently yields hexagonal-TiAlN. On the other hand, synthesizing cubic-TiAlN with x>0.9 was reportedly enabled by chemical vapor deposition (CVD), whose cutting performance is satisfactory for industrial application. However, film thickness distribution was found even in the lab-scale reactor, which is the biggest hurdle for mass production. Computer-aided reactor design is the only possible solution for it; whose reliability is definitely reliant on the quality of kinetic reaction model. Kinetic analysis was therefore carried out for TiAlN-CVD at 800 °C from TiCl4-AlCl3-NH3-H2-He, by changing the partial pressure of each metal chloride and NH3 (PAlCl3, PTiCl4, and PNH3). During the TiAlN growth, growth rate of AlN ingredient increased with PTiCl4, and vice versa, suggesting that some interactive reactions should exist between them. Gas-phase reaction simulation was performed by using existing elementary reaction model, but could not explain this interaction. Thus, individual growths of TiN and AlN were examined at 800 °C and PNH3=19.2Pa. Growth rate proportionally increased with precursor partial pressure as shown in the figure but decreased with the increase of PNH3 for all growths (data is not shown). TiAlN-CVD was previously found to be diffusion limited, but the decrement cannot be rationalized by the change of mass-transport coefficient. It should rather be regarded as transient region of diffusion and reaction limited. We thus performed step coverage analysis to extract surface reaction rate constants, and found that competing Langmuir-Hinshelwood (LH) mechanism could explain the overall phenomena. Consequently, complicated interactive reaction between Ti and Al precursors are getting clarified.
Titanium dioxide (TiO2) is applied to photocatalysts, photochromic materials, solar cells, etc. due to its characteristic electrical, chemical, and optical properties. Recent studies revealed that the photocatalytic and other performances of TiO2 thin films can be improved when silver (Ag) or carbon nanotubes (CNTs) are embedded in the films because of the changes of light absorbance and electron/hole transfer. Such films have usually been synthesized by liquid-based methods. However, the methods require many procedures, and the control of the properties and amount of the embedded nanomaterials is not easy. We recently developed a CVD technique to synthesize thin films in which well-characterized, pre-formed nanomaterials are embedded [1]. In this study, TiO2 thin films with embedded Ag nanoparticles and CNTs are synthesized by applying this technique. The properties and photocatalytic performance of the synthesized films are evaluated.
Titanium alkoxide vapor was fed into a microwave plasma field to form a TiO2 matrix. An aqueous suspension of either or both of Ag nanoparticles and CNTs was sprayed, and the resulting droplets were fed into the plasma field simultaneously. These simultaneous feedings enabled the synthesis of nanocomposite films through a one-step plasma-enhanced CVD process.
Observation of the films using a scanning electron microscope (see images) confirmed that the CNTs were successfully embedded in the film. Elemental analysis and absorption spectrum indicated the enhancement of the absorbance of visible light due to the existence of Ag and CNTs in the film. The films in which either Ag nanoparticles or CNTs were embedded showed higher photocatalytic activity than a TiO2-only film. The film with both of Ag nanoparticles and CNTs exhibited a further higher activity. The film caused 1.8 times dye-decomposing reaction rate of the TiO2-only film under visible light irradiation.
[1] Kubo et al., Thin Soild Films, 632, 55 (2017)

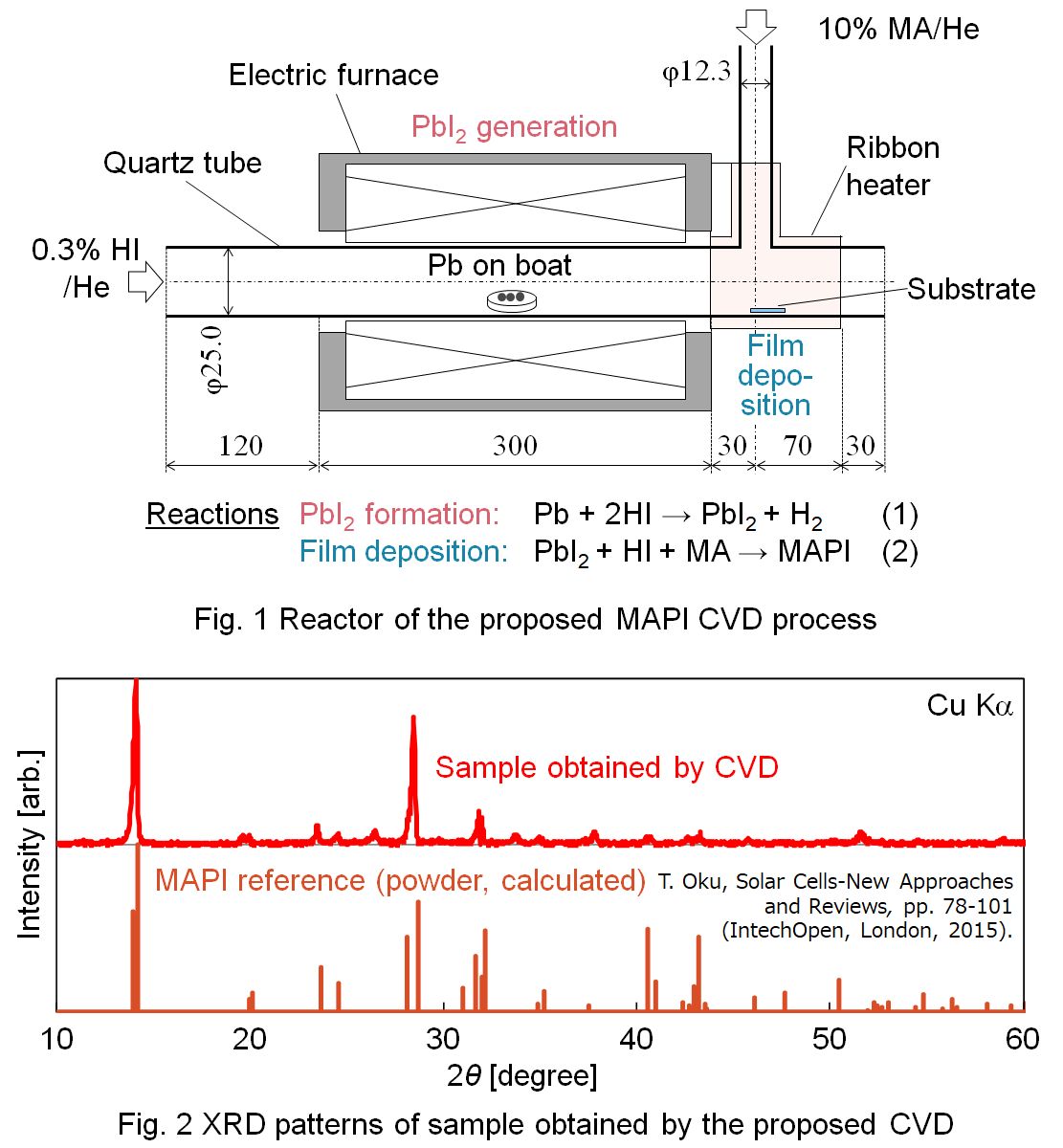
Methylammonium lead iodide (MAPI, CH3NH3PbI3) perovskite solar cells have attracted great expectations as next-generation low-cost solar cells, and development of large-scale film fabrication processes is required. Development of a chemical vapor deposition (CVD) process is required for its capability for large-area and high-purity fabrication of thin films. Although a CVD process for fabricating MAPI was proposed,1) it includes solid reactants that should be sublimated and therefore the flow rate control is difficult. In this study, a novel CVD process to synthesize a MAPI thin film from gaseous reactants, hydrogen iodide (HI), monomethylamine (MA, CH3NH2), and lead (Pb) vapor is proposed. As shown in Fig. 1, the reactor consists of two tubes. Molten Pb was placed in a tube heated at 873 K, and 0.3 % HI in He was supplied for generating PbI2 vapor in Reaction (1). PbI2 and HI were mixed with 10 % MA in He supplied from the other tube, to form a product film on a 10 mm×10 mm quartz substrate heated at 423 K in Reaction (2). The total pressure was approximately 2 kPa. Flat red-brown films were obtained at 423 K for 30, 60, 180 min. The film thickness was 0.4, 1.0, 2.8 μm respectively. The mass of the samples are on the liner plot. XRD patterns and UV-vis spectra of the obtained samples are in good agreement with the reference pattern of MAPI. MAPI was successfully synthesized by a proposed CVD method. XRD patterns of obtained samples show that the PbI2 peak decreased and the MAPI peak increased with time. The result indicates that the MAPI crystal grew with time, which can also be indicated by the result of observation with SEM.
1) M. M. Tavakoli, L. Gu, Y. Gao et al., Sci. Rep., 5, 14083 (2015).
Utilization of CO2 by converting it into useful chemicals is sought. Catalytic hydrogeneration of CO2 to methanol is an attractive pathway as it is an important feedstock. So far, for this reaction, Cu/ZrO2 has been proposed as a promising catalyst and the interface of Cu-ZrO2 is considered as the active site. Therefore, high Cu loading and small Cu particles are essential for maximizing the number of the active site. Here, we successfully produce highly-loaded CuO (60 wt% as Cu) on ZrO2 nanoparticles by flame spray pyrolysis (see the left side of the figure) that can be scaled up to kg/h of the production rate. By decreasing the precursor feed rate, the particle size decreases because of the shorter particle residence time in the flame, which hinders the particle growth (as shown in the figure). Notably, when the catalyst was prepared at the feed rate = 2 mL/min, any Cu species were not detected by XRD indicating that Cu species were present as an amorphous and/or very small particles that are below the detectable limit. This catalyst showed about 5 times higher selectivity to methanol than a commercial Cu/ZnO/Al2O3 catalyst. In contrast, by preparing the catalyst at the higher feed rate (5 and 10 mL/min), CuO peaks appeared in the XRD pattern and their selectivity to methanol is about 50% less than that prepared at 2 mL/min of the feed rate. Therefore, the catalyst prepared at the feed rate = 2 mL/min that consists of smaller CuO and ZrO2 particles than that prepared at the feed rate = 5 and 10 mL/min would be beneficial as they maximize the interface of Cu-ZrO2 sites.
Recently, lignocellulose biomass has attracted as new carbon resources. Separation of the biomass components, which are cellulose, hemicellulose, and lignin, is necessary for biorefinery, and some separation techniques have been developed for long years. Especially, organosolv method, in which a mixture of water and organic solvents is used for depolymerization of lignin and hemicellulose, has been focused on due to its potential for contribution to biorefinery.
Many researchers have investigated kinetics of delignification and pointed out the effect of mass transfer on the delignification rate. However, no one have modeled the rate of delignification including effects of chemical reaction and mass transfer.
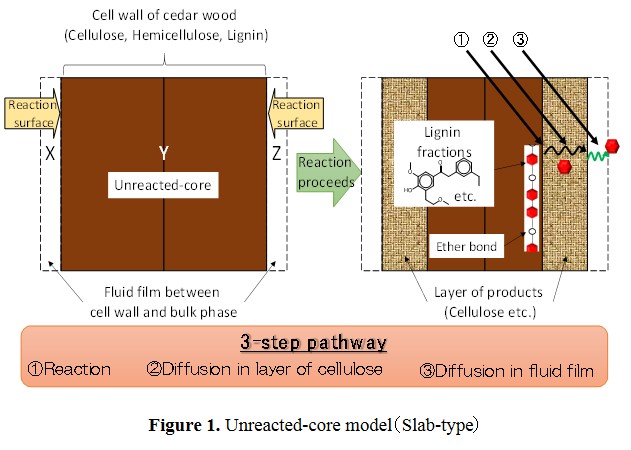
In this study, we have tried to model kinetics of delignification on organosolv method using 1-butanol by unreacted-core model, as shown in Figure 1. This model is used to express situations in which solid particles are being consumed by reactions, and the amount of the material being consumed is shrinking.
Delignification of cedar wood was carried out in a water/1-butanol mixture using batch reactor at 473 K for 0–8 h. After delignification, the products were filtered and divided into liquid and solid products. The solid products were dried, and then analyzed using elemental analyzer to measure carbon content. Chemical composition of cedar wood and the solid products were decided by NREL method.
Conversion of lignin (XL) was calculated in terms of carbon. Based on the unreacted-core model, the overall delignification rate equations were derived for the 3-step pathway shown in Fig.1. The experimental data were found to be well expressed by the rate equation derived on the basis of diffusion in layer of cellulose as the rate controlling step fot the overall delignification rate.
Acknowledgement
This work was supported by the Advanced Low Carbon Technology Research and Development Program (ALCA) as a project from Japan Science and Technology Agency.
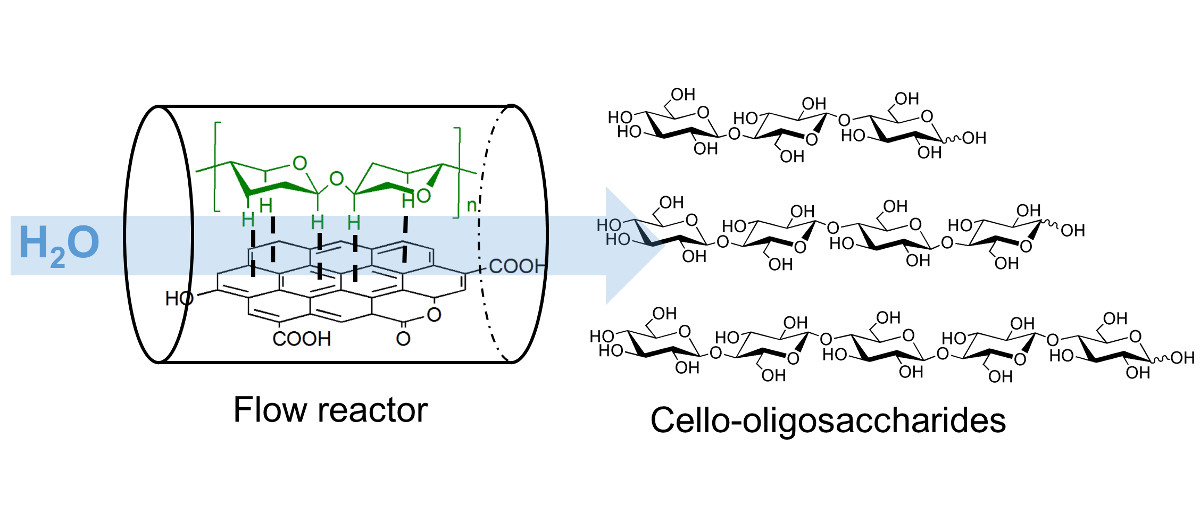
Cello-oligosaccharides are short chain bio-active molecules with the ability to improve the immune defense of plants, animals and humans. These oligosaccharides are only produced in very small quantities and their commercial use is restricted due to the absence of an efficient synthesis process.
Cellulose has a similar structure to these oligosaccharides albeit with a high chain length. Partial hydrolysis of cellulose can produce the desired oligosaccharides but the reaction is difficult to control because the reactivity of molecules increases with the shortening of polymer chain in a conventional hydrolysis reaction.
We have invented a carbon catalyzed semi-flow process for synthesis of food grade cello-oligosaccharides by partial hydrolysis of cellulose. Carbon catalyst was decorated with oxygenated functional groups, which acted as the active sites for hydrolysis. High rate of hydrolysis was achieved by adsorbing the cellulose on carbon catalyst by a solid-state milling step. During reaction, the semi-flow process rapidly removed the dissolved cello-oligosaccharides preventing their further hydrolysis to monomers. Total cello-oligosaccharide yield of 72 % was obtained by optimizing the process at a space velocity of 70 h-1 and a temperature of 473 K.
The composition of oligosaccharides mixture was determined by developing a quantification method based on MALDI-TOF mass spectrometry. The data showed main products had chain length of less than 8 monomer units but oligosaccharide with chain length as high as 13 was also detected. Kinetic data showed the rate of hydrolysis reduced with a reduction in chain length owing to the weaker adsorption of smaller oligosaccharides on carbon surface.
Consequently, we were able to produce a drop-in cello-oligosaccharide solution from cellulose with high yield. The product solution was free from any contamination and could be readily used in healthcare and agriculture industry.
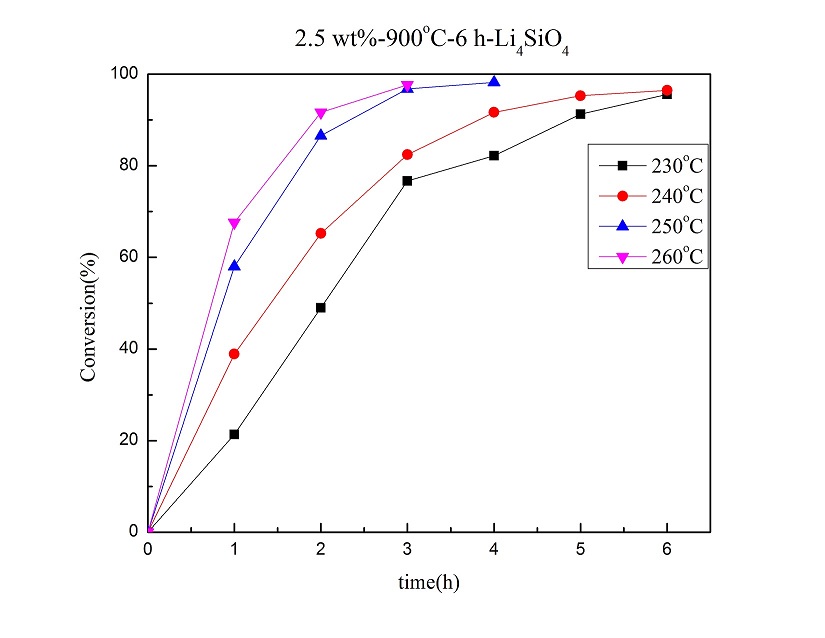
The catalytic solvent-less etherification of glycerol for glycerol oligomers using lithium orthosilicate (Li4SiO4) as a heterogeneous basic catalyst was studied. The Li4SiO4 catalyst was synthesized by solid-state reaction, and characterized mainly using various instruments, including XRD, BET, SEM, ICP-OES, ss-NMR, and the automatic titrator. The etherification reaction was performed from 230 °C to 260 °C for 6 h. GC analysis was applied to quantify the reaction products after being treated properly with silylation. With Li4SiO4 calcined at 900 °C for 6 h, the solvent-less etherification with 2.5 wt% Li4SiO4 catalyst at 240 °C for 3 h resulted in 82% for glycerol conversion and near 87% in the selectivity of di- and tri- glycerol in linear form. According to the ss-NMR and XRD analyses, Li4SiO4 was mostly transformed into lithium metasilicate (Li2SiO3) after reaction. Evidently, a significant portion of Si-O bonds of Li4SiO4 were broken during the glycerol oligomerization. Based on the Eley-Rideal mechanism, the activation energy of this catalytic reaction is 85.93 kJ/mol.
Keywords: Etherification, Glycerol, Lithium Orthosilicate, Catalyst, Diglycerol, Triglycerol
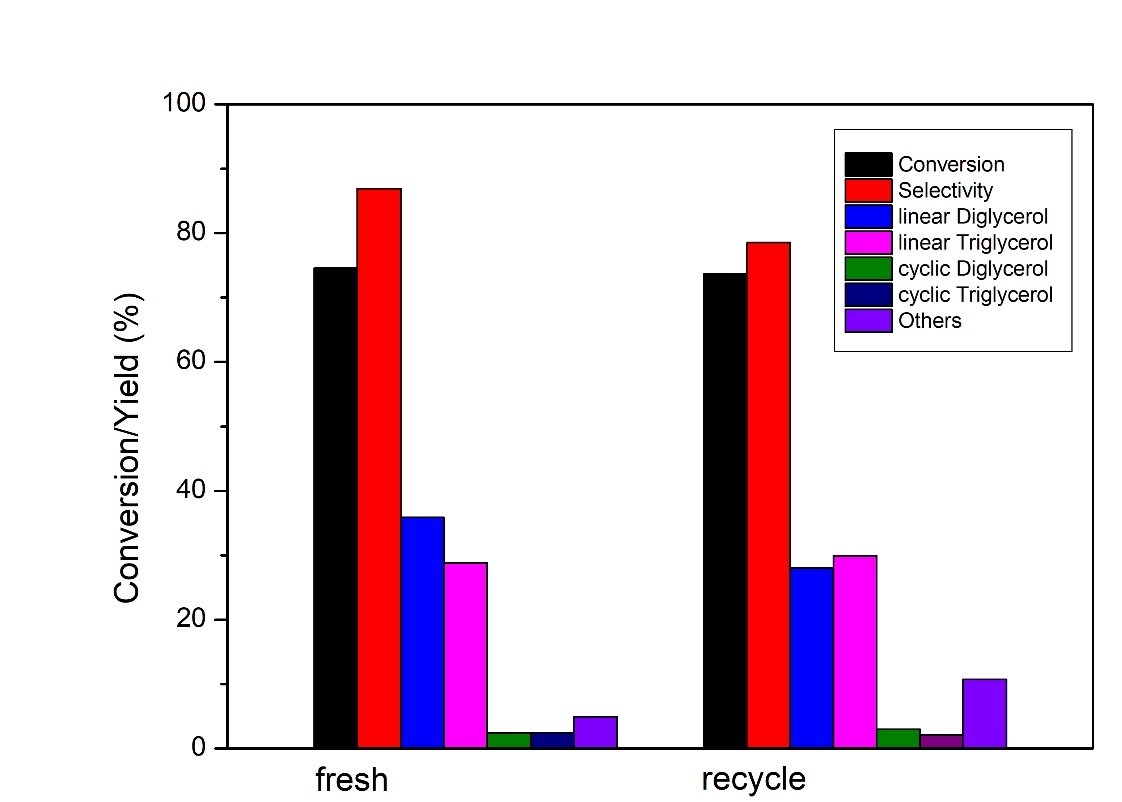
In this work, alumina supported Ca/La mixed oxides as efficient catalysts in oligomerization of glycerol were successfully prepared by the wetness impregnation method using La(NO3) and Ca(NO3) solutions as well as boehmite as support, and followed by calcination at 873K. Various instruments including BET, TPD, XRD, SEM, and ICP-OES analysis was employed for characterization of these catalysts. The catalytic etherification reaction of glycerol was typically conducted at 513 – 553K. In a typical run, the reaction was performed at 553K with a catalyst loading of 3 wt%, based on the mass of glycerol as the sole reactant and the atomic ratio of Ca/La = 2.7 on the catalyst. After 8 hours of catalytic etherification, nearly 75% of the glycerol could be converted to glycerol oligomers, while the selectivity of di-glycerol and tri-glycerol over all products was near 87%. The reusability and stability of the catalyst were tested. It was found that the recovered catalyst still showed good stability and conversion in two consecutive reactions. After 8 hours, the reaction still achieves 78% for glycerol conversion and 75% as the selectivity.
Keywords: etherification; alumina; glycerol; di-glycerol; tri-glycerol; Ca/La/alumina
In this study, Ni/Mo bimetallic catalysts supported on zeolite ZSM-5, aka Ni-Mo/ZSM5, catalysts were successfully prepared by the wetness impregnation method to heterogeneously hydrotreat palmitic acid with an aim for the production of liquid alkane fuels. A Ni-content about 10 wt% of zeolite ZSM-5 support with various Mo-contents was found on these Ni-Mo/ZSM5 catalysts. Various instruments, such as XRD, SEM, BET, XPS, NH3-TPD and H2-TPR, were employed to characterize these catalysts. Typically, the catalyzed hydrodeoxygenation (HDO) reaction of palmitic acid was carried out in a batch autoclave at 573 K with an initial pressure of hydrogen at 35 bar at this temperature. With Ni-Mo/ZSM5 catalysts, the conversion of palmitic acid could reach near 99% at 573K after HDO reaction for 4h. Furthermore, a significant portion of isomerized alkanes were obtained in the product as revealed by GC-MS. That is, both hydrodeoxygenation and hydroisoermization reactions took place conjointly with Ni-Mo/ZSM5 catalysts.
Keywords: Biofuel, Palmitic acid, Zeolite ZSM-5, Ni-Mo/ZSM5 catalyst, Hydrodeoxygenation.
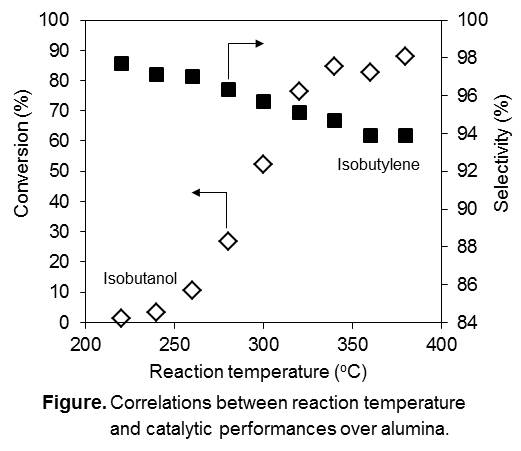
Methyl methacrylate (MMA) has been widely utilized as a raw material of poly methyl methacrylate (PMMA), a typical transparent polymer, and its global manufacturing capacity of MMA is ca. 4,500 kte/y. A few processes have been industrialized so far and ca. 30% of MMA can be produced from isobutylene. In fact, our MMA plants starting from isobutylene are well working in the world. Industrially, isobutylene is mainly derived from naphtha and it is preferable to find alternative isobutylene sources for further expansion of our activity. On the other hand, dehydration of an alcohol is a very useful reaction to obtain the corresponding olefin. As you may know, much attention has recently been paid on isobutanol dehydration to produce isobutylene, especially for the dehydration of bio-based isobutanol into isobutylene, which can be potentially utilized in MMA process. Therefore, our aim is to establish an efficient process for isobutylene production from isobutanol by dehydration. The established dehydration process could be combined with our MMA process starting from isobutylene. We have investigated the dehydration of isobutanol over alumina catalysts. The conversion of isobutanol reached 85% with 95% selectivity of isobutylene at 340oC. The main by-products were linear butenes that formed by isomerization reaction but it was reduced at high concentration of isobutanol. As a result of kinetics studies, it indicated that the reaction pathway consisting of the formation and decomposition of diisobutyl ether became preferable under high concentration of isobutanol. Characterization of alumina was executed to see correlations between crystal structure, acidic properties and catalytic performances. In addition, a long-term continuous reaction at a bench scale showed very stable performances for more than 1,500 h and a design of the potential process flow was proposed.
Industrial metallocene catalysts have to be supported on a MAO modified solid carrier. Micro-sized amorphous and porous silica is the most common used for supporting metallocence / MAO catalysts.
Commercial porous silica is made by a multi-step process with aqueous alkali metal silica as the precursor , which makes it less cost effective and not very environmentally friendly. Thus, there is a need to find a less expensive and more environmentally friendly silica precursor. Silica generated from agricultural waste is more cost effective and environmentally friendly than silica from traditional commercial processes.
In this study, spherical silica particles with a diameter of around 120 nm were fabricated from rice husk ash (RHA), and were used to support two bridged zirconcene complexes ((I) Me2Si(Ind)2ZrCl2 and (II)C2H4(Ind)2ZrCl2 ) for catalyzing propylene polymerization to produce polypropylene (PP) in a temperature range of 40-70 °C and in a solution MAO range of 0.1-0.6 wt%. Due to its small particle size, RHA-supported catalyst exhibited much higher activity than micro-sized commercial silica supported catalyst. At the optimum polymerization temperature of 55 °C and with increasing MAO concentration, polymer yield increased proportionally with the increase of number average molecular weight. Compared to (I), (II) produced more polymer molecules but with much shorter chain length, ascribed to the differences of Zr loading and bridge structure. With increasing polymerization temperature, polymer molecular weight and isotacticity decreased rapidly and resulted in a significant change of PP assembly morphology (shape and size). At 55 °C, (I) produced uniform PP assemblies which had dumbbell-like structure with smooth middle section and two fibrillar ends , while (II) produced spherical PP particles. The width of dumbbell middle part was essentially identical to the Batchelor microscale proposed in turbulent mixing theory.
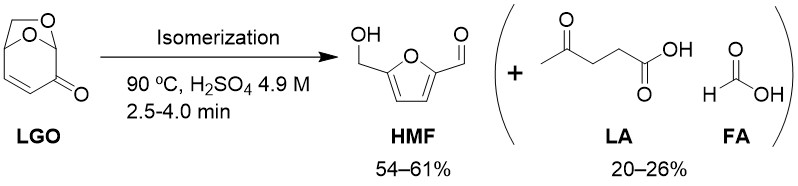
Hydroxymethyl furfural (HMF) is a biorenewable important intermediate molecule for synthesis of a variety of furan derivatives that have a potential in fuel, solvent and polymer applications. Levoglucosenone (LGO), anhydrosugar available from cellulose pyrolysis, has recently been identified as a HMF feedstock. According to the recent studies, LGO can be converted to HMF under milder conditions only with acid and water as catalyst and solvent, respectively. To further explore the potential of this reaction, in this study, we demonstrate the HMF synthesis below 100 °C within a short time of a few minutes at high yields. HMF forms from LGO by the isomerization under acid catalysis, but LGO and HMF suffer from side reactions, excepting the hydration of HMF to levulinic acid (LA) and formic acid (FA), to form humins as unwanted polymeric side products. In the initiation step of this research, we employed high concentration of sulfuric acid (4.9 M) to promote the reaction at low temperatures and carried out the reaction in a batch reactor. However, the yield of HMF was limited to below 30%, and the formation of humins was visually apparent. This was likely caused by the presence of HMF and LGO with high concentration sulfuric acid for a long time to achieve the desired reaction temperature. To solve this problem, the reactor was changed to a microtube reactor, which enabled quick heating of the solution and precise control of the reaction time. As a result, the HMF yield was improved to above 60% in the reaction at 90 °C for 2.5 min. The highest total yield of HMF, LA and FA, indicating the reaction selectivity, was 87% at 4.0 min. When using conventional feedstock such as glucose or fructose, the product yield was much lower even with longer reaction time (<6 min).
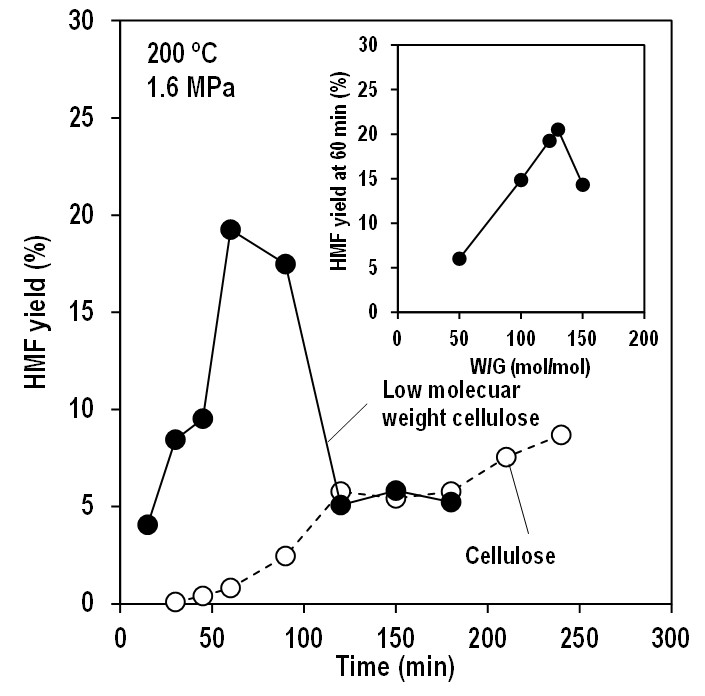
Cellulose is one of the most important biomass resources which can be converted to value-added chemicals, e.g. mono- and oligo-saccharides, furans and organic acids. Among them, hydroxymethylfurfural (HMF), which is precursor of biofuels, polymers, medicine and the other chemicals, is one of the most important target compound. Normally, direct conversion of cellulose to chemicals is difficult because it is stable and insoluble any solvents. Because of these cellulose properties, special solvents and catalysts are developed, however, they are normally expensive and sometime toxic. For these reasons, direct conversion of cellulose to HMF by using eco-friendly way is still challenging research. Here we report that direct conversion of cellulose to HMF using saturated steam, i.e. only water. It is well known that high-pressure, high temperature water, such as subcritical or supercritical water, can promote chemical reactions including hydrolysis and dehydration of saccharides. To directly convert cellulose to HMF, firstly, glyosidic bonds in cellulose must be hydrolyzed, and then, obtained glucose must be dehydrated. Therefore, hydrolysis followed by dehydration must be proceeded in one pot. Because of high hydrolyzability of high-pressure, high-temperature water, cellulose and obtained HMF are easily converted to undesired compounds. To avoid this undesired reactions, we used saturated steam, whose hydrolyzability might be milder than subcritical water. Because of mild hydrolyzability, cellulose was effectively converted to HMF up to 21 %. In addition, we found that pretreatment of cellulose and amount of added water (W/G, molar ratio of water and glucose unit in cellulose) strongly affect to HMF yield. The obtained results indicate that possibility to produce HMF from cellulose using only water, without using special solvents and catalysts.
Lactic acid (LA) is the carboxylic acid that can be applied for using in food, pharmaceutical and chemical industries. An application of using lactic acid as the initial raw material for biodegradable polymer such as polylactic acid (PLA), has been an attractive issue for scientists and engineers. Although lactic acid is commercially produced by fermentation of aqueous glucose using microorganisms, biological processes have various drawbacks for example low reaction rates, and this will lead to the long reaction times and the huge reactors required. The low concentrations of products usually in water will require high energy consumption for separation and purification of lactic acid from water. Therefore chemical processes to synthesis lactic acid from petrochemical resources and other biomasses are still challenged. In this study cellulose, one kind of biomass, is used as the raw material to convert to lactic acid via hydrothermal process in a presence of catalyst. The reaction is conducted in either high pressure reactor or microwave reactor at 200oC, three types of catalysts are used, ZrO2, Al2O3 and mixture of ZrO2 and Al2O3. It is found that the catalytic conversion of cellulose to lactic acid in the high pressure reactor is quite low, the yield obtained from using ZrO2 and Al2O3 are 8.02% and 6.63%, respectively. In the case of hydrothermal process in microwave reactor using ZrO2 as catalyst, it is found that the production yield of lactic acid is higher about 35%. Therefore the microwave reactor can be used as an alternative method to increase the production rate of lactic acid.
In this study, the non-isothermal reaction characteristics and kinetics of biomass devolatilization and char gasification were investigated by TGA. Four kinds of devolatilization atmosphere (N2, N2+CO2 (10%), N2+CO2 (50%), N2+steam (10%)), three kinds of gasification atmosphere (N2+CO2 (10%), N2+CO2 (50%), N2+steam (10%)) and different heating rates (5, 10, 20, 30, 40 K per min) were adopted. Meanwhile, the gasification behaviors of in-situ char and ex-situ char were also compared. The results show that the volume reaction model and shrinking core reaction model can describe well the reaction behavior of biomass devolatilization and char gasification, respectively. With the increase of heating rate for a certain atmosphere, the typical reaction temperatures for both biomass devolatilization and char gasification increased obviously. At a given heating rate, the adopted inert and reactive atmospheres displayed a very weak influence on biomass devolatilization, regardless of reaction behavior and kinetics. However, the devolatilization atmosphere indeed had a remarkable effect on the following char gasification even for the same gasification agent. The in-situ char produced from the atmosphere of N2+steam (10%) had a higher reaction rate and a lower E than the ex-situ char generated from the N2 atmosphere, indicating the necessity of adopting in-situ char for gasification reaction analysis. For the adopted atmospheres, the calculated values of E for char gasification were ranked from large to small as N2+CO2 (50 %), N2+CO2 (10 %),N2+steam-2 (10 %),N2+steam-1 (10 %).
In order to obtain a phosphate rock equivalent, that is, calcium phosphates, from incineration ash of chicken manure which was obtained from power generation system using the manure, the incineration ash was treated with an aqueous solution of nitric acid to elute phosphorus. In using 0.3 M HNO3, most of the phosphorus could be eluted from 1.0 g of the ash within 0.1 h. Compared to composted chicken manure which was previously examined in our laboratory, the concentration of HNO3 should be increased for the elution. However, in case of incineration ash of chicken manure, since it was possible to remove inorganic species having a lower boiling or sublimation temperature and organic species by calcination in the power generation system, the concentration of phosphorus contained in the incineration ash together with the nitric acid extract became higher than those in the composted chicken manure. When nitric acid extract thus obtained was treated using aqueous NH3, the precipitation was formed. XRD showed that the precipitation is poor-crystallized calcium hydroxyapatite (Ca10(PO4)6(OH)2) which is one of main component in the phosphate rock. In order to confirm the certain formation of calcium phosphate species and the purity, the precipitation was calcined at 1,078 K for 5 h. XRD revealed that the calcined solid was tricalcium phosphate without evident contamination. These results revealed that phosphate rock equivalent could be easily obtained from the incineration ash of chicken manure and approximately 14% of phosphate rock imported into Japan can be replaced by the precipitation obtained from of the elution-precipitation treatment of the incineration ash of chicken manure.
Fenton reaction has been believed to be effective for degrading organic pollutants in the wastewater because Fenton reagent, ferrous ion and hydrogen peroxide, is not harmful and expensive. However, it is also well known that oxalic acid, a typical product of Fenton oxidation, forms oxalate complex of ferric ion to interfere with the regeneration of ferric ion to ferrous ion. As a result, Fenton oxidation is quenched with the generation of oxalic acid. Irradiation of UV light is often used to enhance the mineralization utilizing ligand to metal charge transfer of oxalate complex; however, energy consumption of light irradiation is costly. It is desired to accelerate Fenton oxidation to enhance mineralization.
In this study, effect of copper ion on the mineralization of phenol is examined. Mineralization of oxalic acid was enhanced in the presence of copper ion. It is well known that oxalate complex of ferric ion is hard to be decomposed by OH radicals generated by Fenton reaction. This is the reason why that mineralization of oxalic acid is inefficient on the light-assisted Fenton oxidation. In the presence of copper ions, oxalate complex of copper ion is formed. To clarify the reactivity of oxalate complex of copper ion with OH radicals, DMSO was added as a scavenger of OH radicals. Mineralization rate was significantly lowered in the presence of DMSO, indicating that reactivity of oxalate complex of copper ion is rather high. 500 mg/L phenol could be completely mineralized within 120 min in the presence of copper ion whereas more than 5 h is necessary in the absence of copper ion. Significant acceleration of regeneration rate of ferric ion was also observed in the presence of copper ion.
With the rapid and tremendous development of industries in the past few decades, several hazardous pollutants, especially arsenic, selenate, et al, were discharged into aquatic streams. Considering humans' health, the highly detrimental metal or non-metal ions beyond their permissible limits should be removed from the wastewater before discharge. The removal of metal or non-metal species from contaminated water has attracted worldwide attention of scientists. However, some species such as arsenite and selenate are extremely difficult to be removed by conventional methods.
With the aim to find an effective removal method for arsenite from wastewater, we applied supported Pt catalysts onto the removal of arsenite for the first time. The catalytic activity of the Pt catalysts on the removal of arsenite was investigated.
Arsenite was successfully oxidized to arsenate which can be easily removed by conventional precipitation method, with oxygen over supported Pt catalyst in an aqueous solution. The catalytic oxidation of arsenite with oxygen over Pt catalyst obeyed first-order kinetics, and the apparent activation energy was 50 kJ/mol. The catalytic activity of Pt catalyst was significantly influenced by the Pt particle size. The Pt catalyst possesses an excellent stability and can be used repeatedly with a high catalytic performance.
Thus Pt catalyst has a splendid prospective application on the removal of arsenite from contaminated water due to its outstanding catalytic performance, high chemical stability and recyclability.
The automotive three-way catalyst (TWC) is composed of platinum group metals (PGMs), which are mainly deposited on washcoated cordierite ceramic substrates via wet impregnation processes. Another substrate that consists of thermally durable Fe–Cr–Al metal foil is also used due to their attractive features such as higher thermal conductivity and lower pressure drop compared with conventional ceramic substrates, whereas the honeycomb catalysts are prepared via the same wet impregnation and/or slurry coating regardless of the different substrate materials. We recently succeeded to prepare an active Rh nanolayer catalysts on the Fe–Cr–Al metal foils using a pulsed arc-plasma (AP) deposition. Regardless of the very small geometric surface area of metal foils with nonporous and smooth surface, this new catalyst delivered a high catalytic performance for simulated TWC reaction with high turnover frequency compared with conventional Rh powder catalyst (Rh/ZrO2). A miniature honeycomb (11 mm×10 mm, 900 cpsi) was easily fabricated using flat and corrugated metal foils after coating of Rh nanolayer (Fig. b), and it showed steep light-off for NO, CO and C3H6 and complete conversion in the simulated TWC test at GHSV = 120000 h–1. Due to unnecessary of conventional wet processes such as impregnation and washcoating, we successfully fabricated the metal honeycomb converter with much higher cell density of 4,000 cpsi (Figure c) and 9,000 cpsi (Figure d), the activity of which was enhanced. The AP-processed honeycomb catalysts with surface-modified foils are promising not only for automotive TWC applications, but also for other industrial catalysts. It is a simple dry process compared to conventional wet catalyst preparation and thus the substantial decrease of PGM use may also be expected.

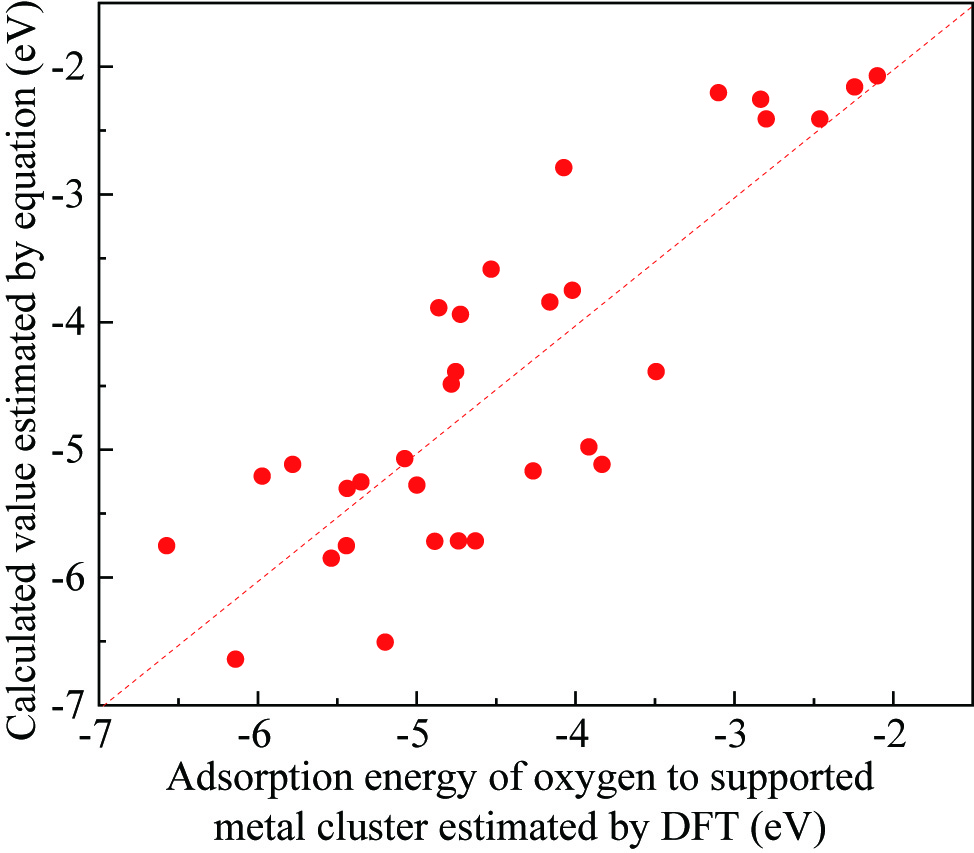
This study carried out the Density Functional Theory (DFT) calculation, to understand the catalytic activities of supported noble metal catalysts. The adsorption energies of the oxygen on the supported noble metal clusters (EDFT) were estimated for several structure models (e.g. Pt3/TiO2, Pt3/Al2O3, Pd3/TiO2, Pd3/Al2O3, Rh3/TiO2, Rh3/Al2O3 etc.). As a result, it was found that the EDFT varies depending on the chemical potential of oxide support (mMOx), d-band center of noble metal (ed) and the number of atoms (n) contained in the structural model. We proposed the empiric formula, Ecal=C1(mMOx-mO)2n/(n+1)+C2ed+C3, to estimate the EDFT values. The unknown parameters, C1, C2 and C3, can be determined so that the Ecal values corresponds the EDFT values. The figure shows the relationship between the Ecal and EDFT values. This correspondence suggests that the EDFT values can be estimated by obtaining the mMOx and ed values by the DFT calculation. The Brønsted–Evans–Polanyi (BEP) principle implies that there is a linear relationship between the activation energies (Ea) of the supported noble metal catalysts and EDFT values. Namely, there is a possibility that the catalytic activity (Ea) can also be estimated by the mMOx and ed values via Ecal values. To verify this possibility, this study carried out the experiments. The supported precious metal catalysts, with different noble metal (Pt, Pd, Rh) and different support oxides (Al2O3, SiO2, TiO2 etc.), were prepared by the impregnation method. Then, the propylene (C3H6) oxidation activities for prepared catalysts were analyzed by a fixed bed flow reactor. Moreover, the Ea values were obtained by the Arrhenius plot. As a result, a well matched correspondence was found between the Ecal and Ea values.
A photoresponsive tracer that changed color under ultraviolet (UV) irradiation was synthesized by immobilizing a photoacid generator and methyl orange on a poly(vinyl chloride) (PVC) carrier resin to understand photoreaction behavior in a dry powder photoreactor. The tracer changed color as a function of the received cumulative radiant energy.Tracer particles were fluidized in a fluidized bed photoreactor under various conditions and exposed to UV irradiation from the side of the reactor. The color change of the tracer during irradiation was recorded using a video camera. By analyzing the recorded images, the color transition of the tracer could be continuously detected as a change in the green component of RGB color value. The apparent photoreaction rate constant of the tracer in the fluidized bed photoreactor was successfully estimated by analyzing the rate of change in the green RGB value during irradiation. The transmission of UV into the powder layer were found to obey the Lambert-Beer law. The photoreaction behavior in the reactor was successfully reproduced by a simple two-phase model consisting of a reaction zone and a non-reaction zone. The present method for evaluating the performance of dry powder photoreactors by use of a photoresponsive tracer is very simple. Since the reaction behavior can be visualized, it is intuitively easy to understand and useful for designing dry powder photoreactors.

Advances in device technology have been accompanied by the development of new types of materials and device fabrication methods. Considering device design, initiated chemical vapor deposition (iCVD) inspires innovation as a platform technology that extends the application range of a material or device. iCVD serves as a versatile tool for surface modification using functional thin film. The building of polymeric thin films from vapor phase monomers is highly desirable for the surface modification of thermally sensitive substrates. The precise control of film thickness can be achieved in iCVD, creating a conformal coating on nano‐, and micro‐structured substrates such as membranes and microfluidics. iCVD allows for the deposition of polymer thin films with a variety of chemical functionality, and thus, substrate surfaces can be functionalized directly from the iCVD polymer film or can selectively gain functionality through chemical reactions between functional groups on the substrate and other reactive molecules. These beneficial aspects of iCVD can spur breakthroughs in device fabrication based on the deposition of robust and functional polymer thin films. This presentation reviews recent progress made in iCVD‐based technologies in various fields, especially for soft electronic device applications.
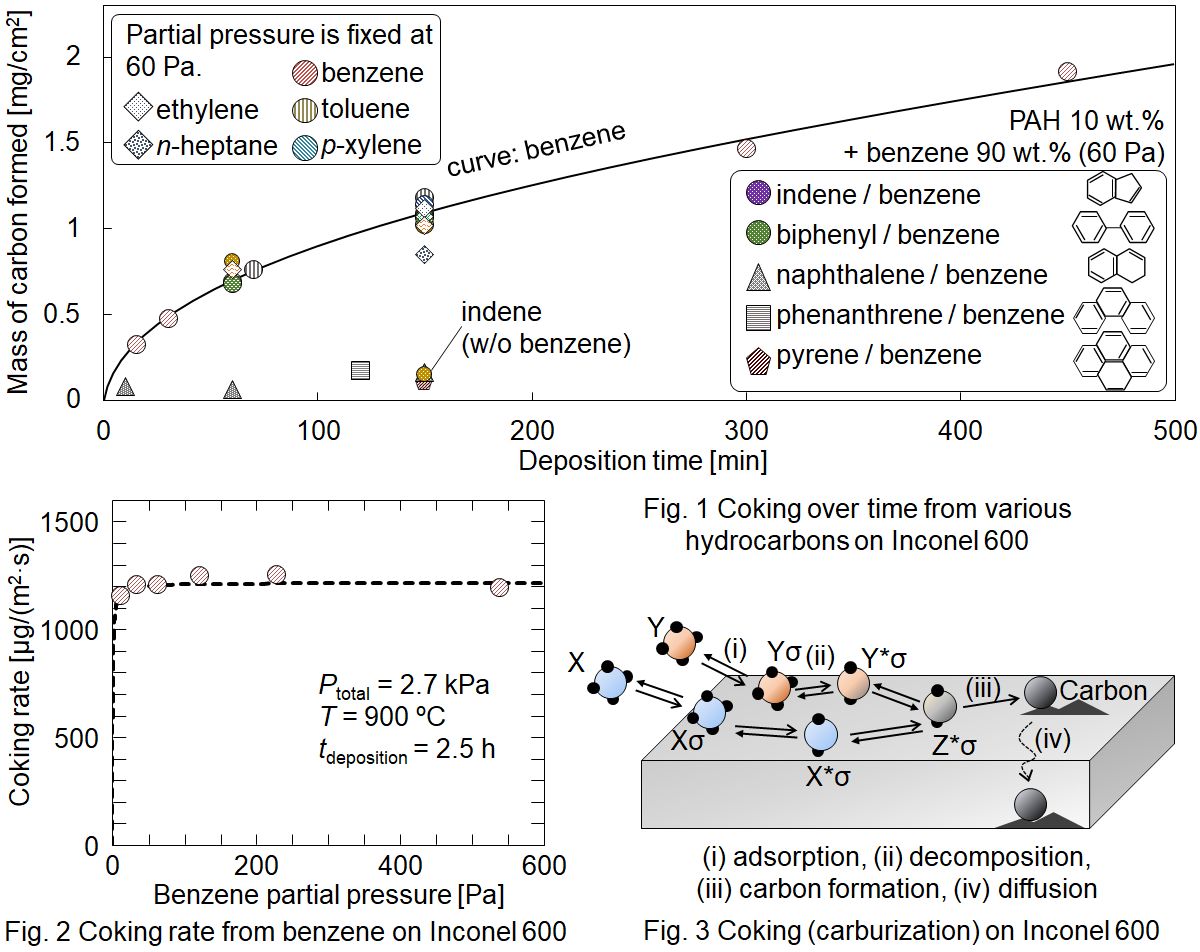
Unfavorable deposition of carbon called coke takes place in metal tube reactors used in hydrocarbon processes in the industry. The coke formation mechanism is unclear. In this study, a laboratory-scale thin tube reactor is employed for investigating mechanism of the coke formation from hydrocarbons on metal and inert substrates. Chemical vapor deposition (CVD) experiments were carried out using a quartz tubular reactor, in which 10 mm×10 mm substrates were placed. One of several hydrocarbons or a benzene solution of one of heavy hydrocarbons was supplied with diluent He to the reactor. The reaction temperature was 900 °C and the total pressure was 2.7 kPa. The space time from the inlet to the substrate was fixed at 15 ms. When benzene was supplied, coking rate was highest on the SUS 316L substrate followed by Incoloy, γ-Fe, Inconel, Nichrome, SUS 430, Ni, Cr, and α-Fe in order. This shows the catalytic activity of respective metals. On the Inconel substrate, the coking rate was approximately equal from ethylene, n-pentane, n-heptane, benzene, toluene, p-xylene, and biphenyl. The rate was almost independent of the reactant partial pressure. These resulted from solid-phase diffusion of carbon which was generated from hydrocarbons adsorbed on the metal surface near saturation. Naphthalene, phenanthrene, and pyrene exhibited ca. 10 times lower coking rate than the other hydrocarbons, which suggests more stable adsorption and slow decomposition on the metal surface. The coking rate on inert substrates, quartz, silicon, and carbon, was ca. 40 times lower than that on Inconel. The rate per unit surface area of the inert substrates obeyed an identical Langmuir-type rate equation. As a result, coking on metal surface starts with adsorption, fast decomposition on the surface, followed by fast carbon diffusion into the substrate, and slow carbon CVD on the deactivated surface continues even after completing the carburization.
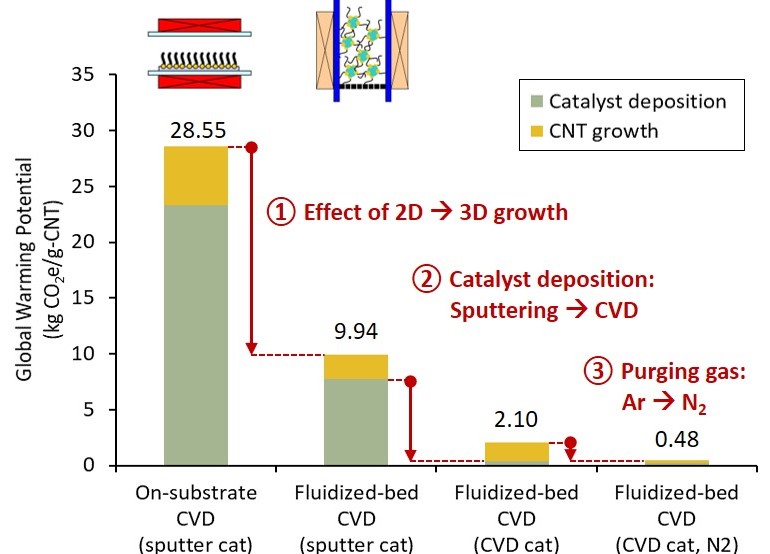
Carbon nanotube (CNT) is an emerging material with promising applications due to its superior properties. Mass production of CNT is most feasible with catalytic chemical vapor deposition (CVD). To synthesize high-quality single-wall to few-wall CNTs, catalyst nanoparticles must first be deposited on substrates. Then, using a high-temperature CVD reactor, hydrocarbons are decomposed into CNT on the substrates. The processes are intensive in material and energy. Yet, the consequential impact on the environment is rarely studied because of uncertainties in syntheses. Leveraging the experience in lab syntheses, this study aimed to evaluate the prospective environmental impact of CNT synthesized via three selected catalytic CVD methods. First, the CO2-assisted growth on flat substrates (on-substrate CVD) [Carbon, 136 (2018): p.143-149], represented the established millimeter long CNT growth. Second, the fluidized-bed growth on spherical beads (fluidized-bed CVD) with sputtered catalysts [Carbon, 50(4) (2012): p. 1538-1545], contrasted the effect of 3D CNT growth to previous 2D growth. Third, the fluidized-bed CVD with chemical vapor deposited catalysts [Carbon, 49(6) (2011): p. 1972-1979], showed a repeatable single-reactor production. We quantified the global warming potential impact of 1 g CNT production with life cycle assessment (LCA). The results for the three syntheses were 28.55, 9.94, and 2.10 kg-CO2equiv/g-CNT, accordingly (illustrated in graphical abstract). By comparing the three syntheses, we showed that the strikingly high impacts were mitigated through increasing CNT yield with fluidized bed and replacing the energy-intensive sputtering process. A scenario to substitute the remaining hotspot, argon (purging gas), with nitrogen could reduce the impact to 0.48 kg-CO2equiv/g-CNT. When considering scale-up from lab-scale (reactor diameter of 22 mm, productivity of 5 g per day) to industrial scale (diameter >30 times, productivity >3 to 104 times), the CNT production will be comparable to industrialized carbon fiber production (about 0.02 kg-CO2equiv/g-CF).
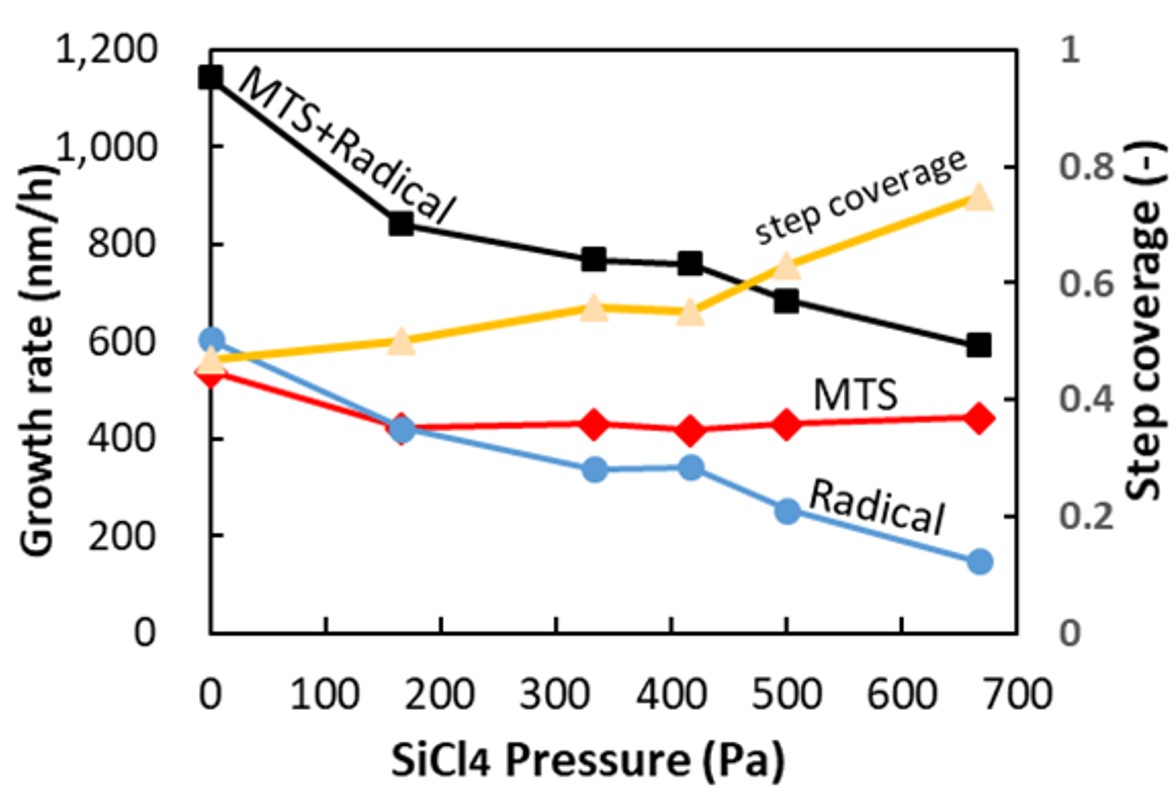
Ceramic matrix composites (CMC) consisting of SiC fibers (SiCf) and SiC matrix receive attention as a material for next-generation aerospace engines due to light weight and high heat resistance. SiCf/SiC-CMC is fabricated by weaving the SiC fibers, followed by infiltrating SiC into their interstices. Chemical vapor infiltration (CVI) from methyltrichlorosilane (MTS) and H2 is employed to infiltrate SiC fibers due to high conformality. However, perfect infiltration was unfeasible so far even by CVI because aspect ratio of the interstice is huge. We have found that SiC was formed by two growth species, one is MTS (contributes to uniformity) and the other is radical species (gas phase decomposition product of MTS, contributes to non-uniformity). Therefore, elimination of the radical species is mandatory for conformal deposition, and we proposed to use sacrificial layer for it [1]. We therefore introduced high MTS concentration (>1.2kPa), to realize higher conformality without compromising growth rate. Based on Langmuir-Hinshelwood (L-H) mechanism, we can achieve quasi-0th-order reaction at high concentration which will contribute to uniform deposition. Step coverage by MTS was thus improved from 60% to 88% using high MTS concentration. We also investigated SiCl4 gas addition into SiC-CVI for scavenging the radical growth species. Figure shows SiC growth rate and step coverage dependency on SiCl4 pressure. When SiCl4 was added, the radical was successfully scavenged, whose impact was enriched with higher SiCl4 concentration, while no impact on deposition by MTS. Consequently, combination of high MTS concentration to achieve L-H mechanism and SiCl4 addition to scavenge radical growth species is the key to achieve high growth rate and high conformality simultaneously.
[1]K. Shima et al., Society for Chemical Engineers Japan 47th Autumn meeting, Hokkaido University, O302 (2015).
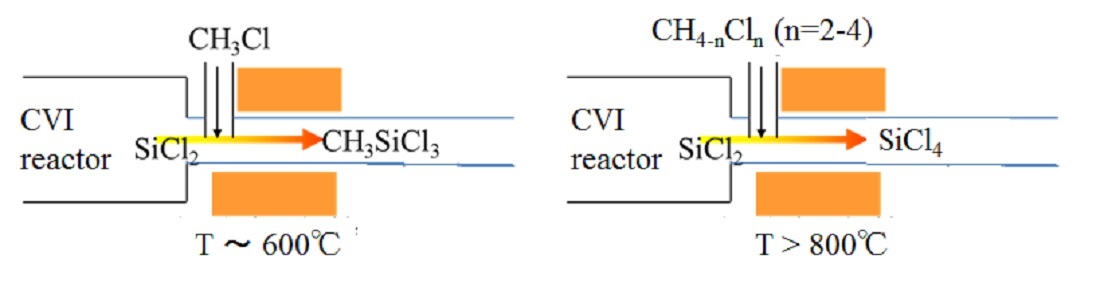
Silicon carbide (SiC) is widely used for structural materials due to high mechanical strength and corrosion resistance. Chemical vapor infiltration (CVI) from methyltrichlorosilane (MTS) is commonly adopted for its conformal deposition capability and high productivity. However, hazardous by-products, polymerized SiCl2 such as (SiCl2)n and X-(SiCl2)n-Y, are known to be piled up in a exhaust system of reactor. It compels a periodic cleaning work that takes several days for mass-productive reactor. To make matters worse, these are stable in vacuum i.e. reactor, while explode via hydrolysis when being exposed to air. Accordingly, we have to develop the way to inhibit (SiCl2)n production without resorting to unsafe experiments. First, the reaction mechanism to form (SiCl2)n was studied theoretically. The rate constants of elementary reactions starting from SiCl2 to (SiCl2)n and Cl-(SiCl2)n-Cl (n=1-3) were calculated with quantum chemical calculations, followed by coupling with existing model for H/C/Si/Cl system. Reaction kinetic simulation using the updated model revealed that SiCl2 is stable at a process temperature, typically above 900 °C; meanwhile, during temperature drop in the exhaust system, SiCl2 reacts into SiHCl3 and SiCl4 at 500-800 °C and (SiCl2)n below 400 °C. As the reaction mechanism was clarified, practical way to inhibit (SiCl2)n generation was sought. Here, we propose to inject additive gas into the exhaust system, which reacts with SiCl2 to form stable species. When CH3Cl is added at 600 °C, residual ratio of SiCl2 decreased to 0.1% in 2 sec (product: MTS). When CHnCl4-n (n=2-4) is added above 800 °C, the residual ratio decreased below 1% in 1 sec (product: SiCl4). It is recommended to add CH3Cl around 600 °C or CHnCl4-n (n=2-4) above 800 °C into exhaust gas to inhibit (SiCl2)n production. This conclusion has already been verified experimentally. Although this work was conducted for SiC-CVI, the same solution is applicable to Si-CVD from SiHCl3 or SiH2Cl2.
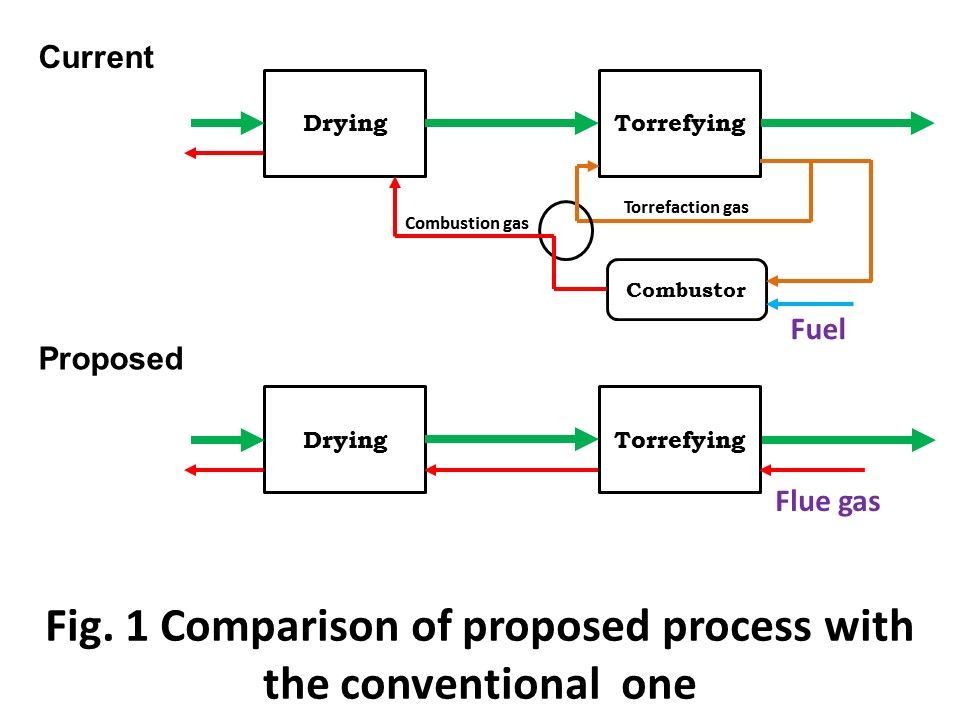
Malaysia is the second largest producer of palm oil, producing 17.3 million tons, or approximately one third of the total world supply in 2016. In parallel with the palm oil production, a huge amount of biomass residues are generated by the industry including oil palm plantations and palm oil mills: empty fruit bunch (EFB) 18.3, mesocarp fiber 11.0, palm kernel shell (PKS) 4.6, trunk 17.4 and fronds 125.3 t-wet/year. Mesocarp fiber is almost fully utilized as solid fuel for producing electricity and steam at palm oil mills. About PKS, some are utilized for electricity and steam at palm oil mills (the same use as mesocarp fiber), some are being traded as solid fuel. EFB is underutilized. There are three representative thermochemical conversion techniques developed for this kind of lignocelluosic biomass residue: torrefaction, fast pyrolysis and gasification, which produce solid fuel, bio oil and syngas, respectively. The most significant obstacle for commercialization of biomass use is not the level of conversion technology itself, but logistics. Logistics for biomass is still really immature in comparison with fossil fuels; such as coal, crude oil and natural gas. In this sense, biomass use in small scale is prioritized way to start due to its lower risk in terms of logistics. In this talk, the presenter will introduce torrefaction , of which product can be commercialized in small scale, or with less logistic risks. Combustion gas utilization for torrefaction will be introduced as a lower running cost process. The concept is illustrated in Fig. 1.
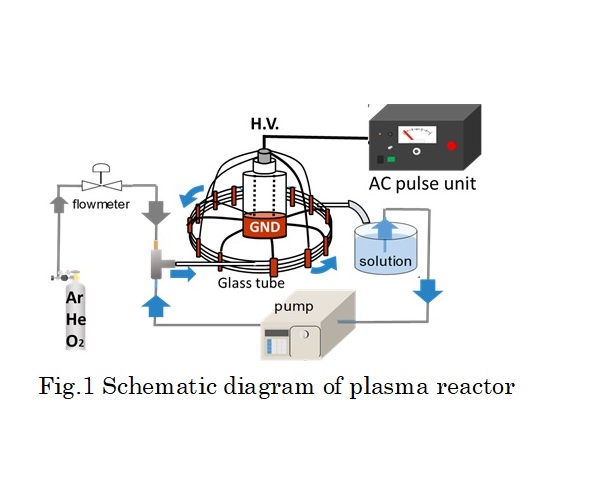
In this study, non-equilibrium atmospheric pressure pulsed discharge plasma technique using a gas-liquid slug flow in glass column is applied to water containing methylene blue (MB) degradation. The reactor was comprised of 4 rows glass column bundled by copper foils that attached on the outside of the column surface. Moreover, the foils were used as high-voltage electrodes totally in 5 parallels. As the ground electrode, copper foils were attached on column at distant of 25 mm from high-voltage electrode in the same way. The water solution and the gas were flowed simultaneously in glass column to form the slug flow system. The generated bubble motion interval was controlled by adjusting the flow rate of gas and liquid. In this time, MB solution and each gas were flowed at 1.5 and 0.15 ml/min, respectively. After the flowing bubbles in the glass column became steady, an electrical discharge was introduced into the slug flow system by using AC power supply with a bipolar pulsed output voltage to generate plasma in bubbles between electrodes. The output voltage is 9 kV at 10 kHz repetition and its pulse duration is 10 μs. The reaction was conducted totally in 120 min, and the degradation ratio was measured every 20 minute. As a result, over 90 % of MB were degraded for all of the gas species (O2, Ar, He). Especially in the case of O2, mostly all of the MB was degraded in 120 min. This implies many kinds of active species originated by oxygen were dissolved in plasma treated water. However, the amount of hydrogen peroxide was higher in Ar and He. Finally it was confirmed that the degradation rate in this method was higher than that in other plasma methods such as atmospheric pressure plasma jet.
With record heat spreads all over the world, global warming has become an issue that cannot be ignored for human beings, and greenhouse gas emissions must be curbed and reduced. In particular, carbon dioxide (CO2), which accounts for the majority of greenhouse gas emissions, is the most important target of emission control and reduction. Although CO2 is a very stable compound with high standard redox potential, it can be converted to industrially useful substances such as carbon monoxide, formic acid and hydrocarbons reduction. It is possible to reduce not only greenhouse gases but also produce a resource. Various studies have been conducted to reduce CO2 using renewable energy, excess thermal energy and electrical energy. We have tried to reduce CO2 electrochemically using metal catalyst electrode and convert it to useful substance. Many studies have revealed that the main products of CO2 electrochemical reduction differ depending on the metal species. In the metal species, this study focused on zinc(Zn). Zn is known to produce carbon monoxide (CO) as a main product of CO2 electrochemical reduction as well as gold and silver, while having abundant reserves. In addition, it is expected to selectively generate CO because the overpotential of hydrogen generation is also large and hydrogen generation is suppressed. In this report, surface modification of Zn electrode has been performed by anodization. By changing the conditions of anodic oxidation, different surface modifications were successfully performed and it was successful that prepared Zn electrodes showed quite different product selectivity such as CO and formic acid in CO2 electrochemical reduction. In order to clarify the factor of this product selectivity, it carried out by surface analysis and electrochemical characteristics of the electrode. It was revealed that the product selectivity can be controlled by changing the anodization condition.
Recent manufacturing processes for high-performance crystal particles require the advanced control technologies of crystal structure and morphology. In order to develop the crystallization process for the desired crystal particles, it is important to understand the crystallization behavior from a microscopic viewpoint and actively control the nucleation and crystal growth in solution. We have studied the crystallization behavior of aqueous microdroplet of salt solution using electrodynamic balance (EDB), where a charged droplet could be electrically levitated in space over the periods of hours. It enabled us in situ observation of crystallization behavior in a levitated microdroplet with enough spatial and time resolutions. In this presentation, we directly observed the nucleation and crystal growth in the aqueous NaCl microdroplet . It was found that the finally produced crystal particles had different several morphologies which were mainly influenced by the diameter and the charge of a levitated droplet.
In essence, ionic liquids (ILs) are salts with melting points at or below 100 °C, which are composed of organic cations and organic or inorganic anions. Owing to their distinctive properties, such as low melting point, wide liquid range, extremely low vapor pressure, sound thermal stability and wide electrochemical windows etc., ILs are used in a number of application areas. However, some recent studies are noteworthy, showing that ILs are flammable during thermal upsets. Other studies have also indicated that the gas phase of decomposition products from ILs is spontaneous combustion and easy ignition. To accomplish the objective, thermogravimetric is used to examine the thermal stability of 1-ethyl-2,3-dimethylimidazolium nitrate ([C2mmim][NO3]; C7H13N3O3). Under the dynamic heating conditions, [C2mmim][NO3] is considered to be decomposed until approximately 280.0 °C. Meanwhile, lower hydrophilicity leads to better thermal stability of [C2mmim][NO3] than [C2mmim][X] (X represents halogen). However, due to the limitations of the dynamic heating experiment, [C2mmim][NO3] is found to be decomposed before the lowest measured decomposition temperature under dynamic conditions. Moreover, the combination of the homemade combustion test device and high speed camera are used to investigate the combustion process of [C2mmim][NO3]. The results indicate that the flammability of [C2mmim][NO3] is caused by the combustible decomposition products (see Figure). Therefore, the entire decomposition and combustion process of [C2mmim][NO3] is fully characterized based on the results of thermogravimetric with fourier transform infrared spectrometer.
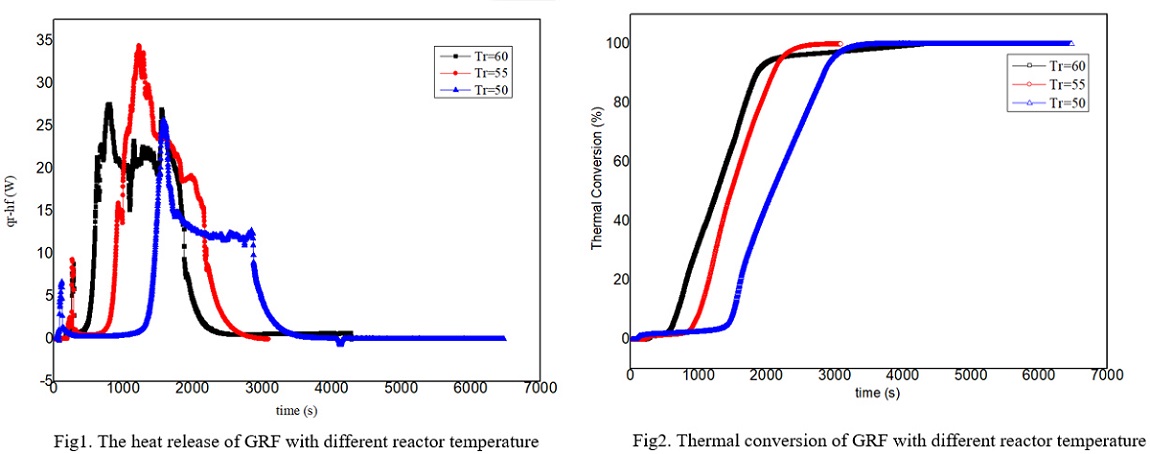
Grignard reagent is often synthesized from magnesium and halohydrocarbon in dry ether such as tetrahydrofuran(THF) or diethyl ether(DE). The Grignard reagent formation(GRF) is a strong exothermic process which has an induction period. If the material is accumulated due to improper initiation, it is easy to cause the thermal runaway after the reaction is suddenly triggered. With the development of green chemistry, more and more chemical companies have begun to develop safer Grignard reagents by changing the solvents. Therefore, it is particularly important to carry out the reaction thermal safety risk assessment for the GRF in different solvents and establish the critical criterion for the thermal runaway reaction of the GRF.
In this work, the n-butylmagnesium bromide GRF was taken as an example. The safety risk assessment of the GRF was carried out by using a cooling failure model using THF, DE, cyclopentyl methyl ether(CPME) and diethylene glycol dibutyl ether(DGDE) as solvents. RC1e and EasyMax have been manufactured by METTLER TOLEDO and used to measure the exothermic conditions of reaction. The kinetic model of GRF was obtained by the experiments. The experimental results show that the safety risk level of GRF by using THF and DE as solvent is grade 3, and it is reduced to grade 1 by using CPME and DGDE as solvent. The latter two reactions have lower exothermic and adiabatic temperature rise than the former two. The critical criterion for the thermal runaway of GRF with THF and DE was established, which has certain guiding significance for the safety of GRF. The experimental results also show that the reaction temperature and the stirring speed have influence on the induction period. In the next step, the authors intend to explore the mechanism of the factors which have the influence on the induction period of Grignard reagent.
Introduction
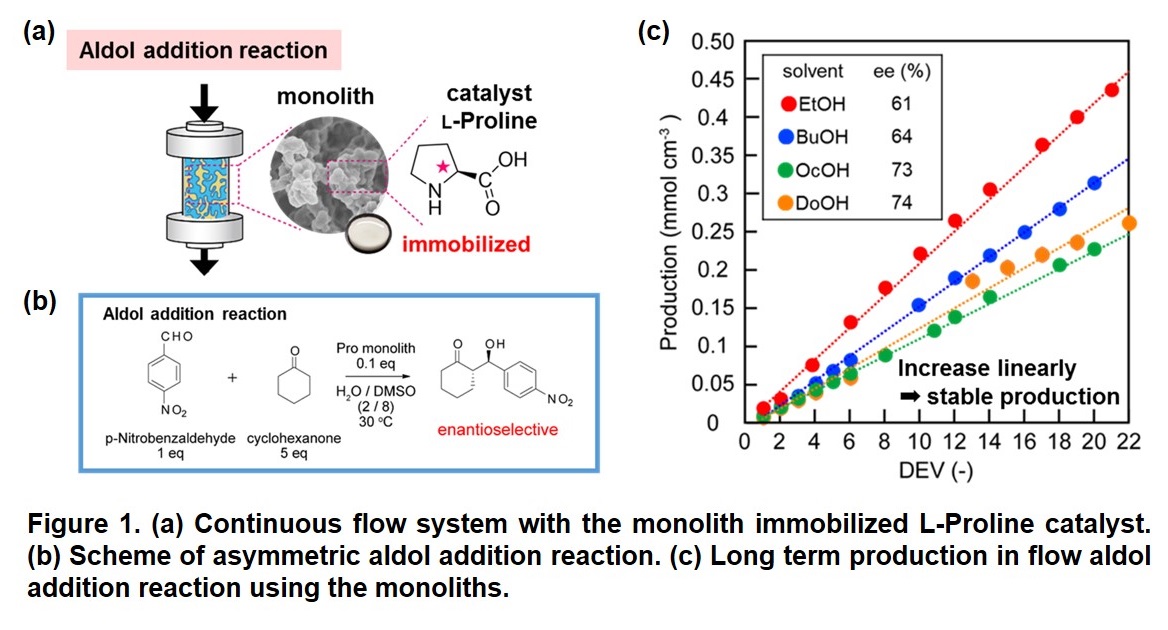
Chemical manufacturing processes in flow system have attracted attention in the field of organic synthesis. To realize energy- and cost-saving processes in the organic synthesis, using immobilized catalyst in flow reactor is a strong tool. Macroporous monolith has a three-dimensional through pore and large specific surface area contributing to its high performance in flow application. In this study, the macroporous monoliths containing L-proline-based organocatalyst were developed for asymmetric aldol addition reaction in flow system (Figure 1 (a)). The effects of porous properties of the monoliths on their catalytic activities and enantioselectivities were investigated in detail.
Results and Discussion
Macroporous monolith was synthesized by copolymerization of O-acryloyl-4-trans-L-hydroxyproline, acrylamide, and N,N'-methylenebisacrylamide in DMSO/alcohol. The porous structures of the monoliths were controlled using different porogenic alcohol (EtOH, BuOH, OcOH, and DoOH). Different length of alkyl chain of porogenic alcohol gave different porous properties, which can be explained by solubility of monomers and polymers in the solvent system.
The L-proline-containing monolith was applied to batch and flow aldol addition reaction between p-nitrobenzaldehyde and cyclohexanone (Figure 1 (b)). Interestingly, the catalytic performance of the monoliths was controlled by their porous properties in batch system. These monoliths with different porous structures were packed in stainless steel columns to construct flow reactors. With a residence time of 12 h, the flow system using the monolith was stable during over 10 days operation and achieved high enantiometric excess (ee) (Figure 1 (c)). However, the porous structure of the monoliths did not affect the catalytic activity and selectivity significantly. To reveal the relation between the monolithic structure and activity and selectivity of the catalytic reaction, the behavior of the substrate solution diffusing through the monoliths will be reported in the presentation.
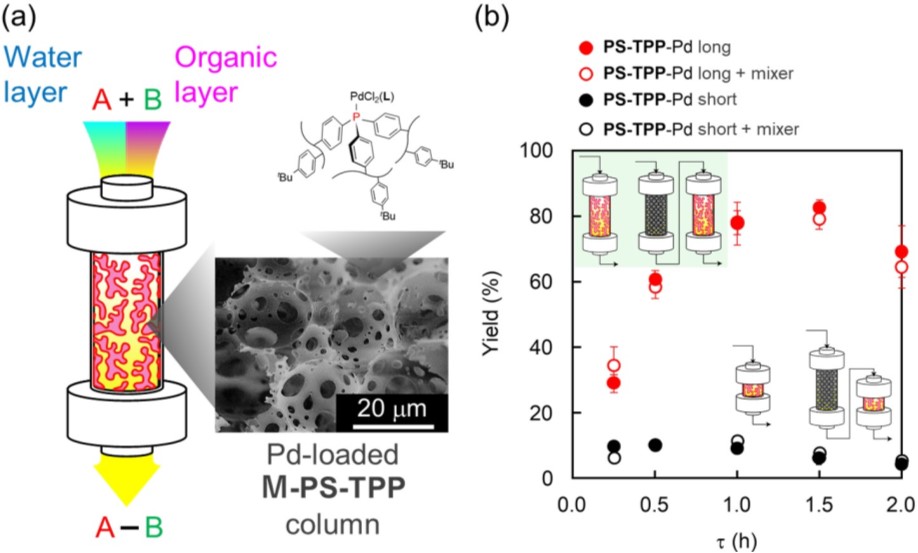
Transition-metal-catalyzed cross-coupling reactions are strong tools for production of active pharmaceutical ingredients with high reaction efficiency, selectivity, and energy saving. Because flow system has advantages over batch system for cost- or energy-efficiency, safety, and automation, the development of flow cross-coupling reaction has been motivated. Particularly, transformation of inactive and inexpensive aryl chlorides under flow condition are highly desirable but have been rarely reported.
Typically, flow cross-coupling reactions require careful design of reactor because of inorganic base reagents and salt byproducts. The inorganic components insoluble in organic solvent cause serious issues of clogging in the reactor. One general solution is to use solvent system which solubilize both organic and inorganic compounds. Some reports have demonstrated that use of organic/water biphasic solvent is beneficial for complete solubility of all the components and efficient cross-coupling reaction. We postulated three-keys to achieve challenging flow cross-coupling reaction: 1) organic/water solvent, 2) immobilized catalyst, 3) efficient mixing; but never demonstrated experimentally yet.
Herein, we first report an application of macroporous polystyrene-triphenylphosphine monolith (M-PS-TPP) to palladium (Pd)-catalyzed Suzuki-Miyaura cross-coupling reaction in flow system (Figure 1a). The M-PS-TPP was synthesized by copolymerization of three-fold cross-linking triphenylphosphine, divinylbenzene, and p-tert-butylstyrene. The macroporous structure of M-PS-TPP was fabricated via polymerization of water-in-oil high internal phase emulsion (HIPE) template. The M-PS-TPP had characteristic window and void structure with the pore sizes of 2–5 and 10–20 micrometer (Figure 1a, inset). Pd-loaded M-PS-TPP column was applied to flow Suzuki-Miyaura cross-coupling reaction between 4-chlorotoluene and phenylboronic acid in THF/water biphasic media containing K3PO4. Tuning the column length and residence time (tau) achieved efficient mixing and high production efficiency in the reaction (Figure 1b).
Silver nanoparticles have caught a wide attention due to its unique physical and chemical properties. Silver nanoparticles embedded in polyvinyl alcohol-polyvinylpyrrolidone (PVA-PVP/Ag) free-standing films are prepared by microwave irradiation within few minutes with varying ratios of the polymers. PVA and PVP perform as reducing agents and stabilizing agents as well as support for silver nanoparticles. The catalyst is characterized by UV-Vis spectrometry, SEM and TEM where the analytical results affirm the reduction of silver ion to silver nanoparticles in the polymer matrix. Good dispersion of silver nanoparticles in the film is confirmed by TEM analysis and the size of silver nanoparticles is found to be in the range of 2-10 nm. This film is used as catalyst for the synthesis of a β-enaminone, 4-phenyl-amino-pent-3-in-one from acetyl acetone and aniline, where silver nanoparticles act well as the catalytic agent of the composite catalyst. A typical reaction shows 89% conversion of aniline to β-enaminone at 600C with this catalyst. Parametric effects on the reaction are studied and heterogeneous kinetic model is proposed for the reaction, where Eley-Rideal model shows good fitting of experimental data. The model parameters are also evaluated and found satisfactory with a high value of correlation factor.
In industrial Vinyl chloride monomer (VCM) manufacturing, the oxychlorination process is most important in terms of elemental efficiency. In this study, we report the reaction kinetics analysis of deactivated catalysts in Oxygen-based oxychlorination plant.
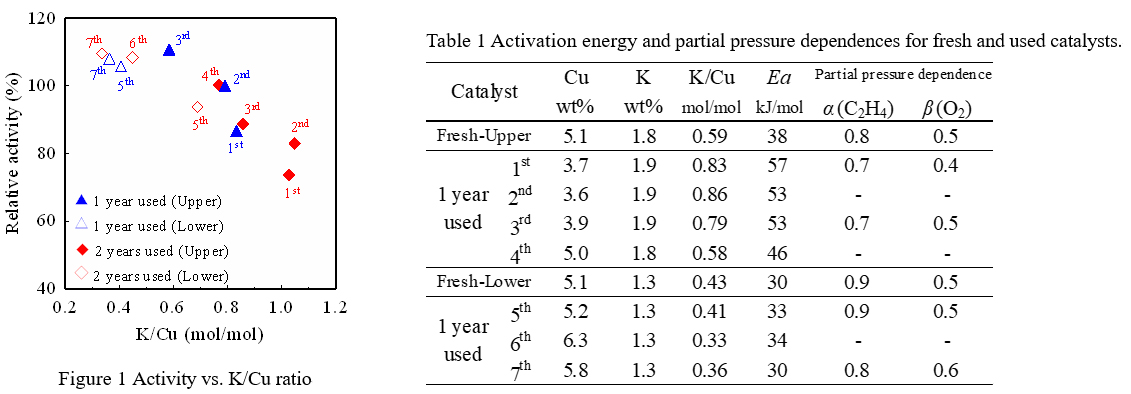
The 11wt%CuCl2-3.4wt%KCl/Al2O3 (Upper) and 11wt%CuCl2-2.7wt%KCl/Al2O3 (Lower) catalysts were prepared according to the conventional impregnation method. These fresh catalysts were subjected to an endurance test in a commercial plant. After two years operation, the catalyst layer was divided into seven positions from the inlet and sampled (1st to 4th were upper catalyst, 5th to 7th were lower catalyst). The chemical properties of fresh and used catalysts were evaluated using a fixed-bed reactor and some instruments.
The figure 1 shows a relationship between oxychlorination activity of the catalyst sampled from each position of the reactor and the ratio of potassium and copper elements (K/Cu). The activity shown here is the relative activity when the initial catalyst performance is 100%. In the used catalysts, the K/Cu ratio increased with operation time due to a decrease of copper accompanying sublimation. As a result, a tendency of activity decreasing due to an increase in the K/Cu ratio was confirmed.
The table 1 shows the activation energy and the partial pressure dependences of raw material gases of the fresh and used catalysts, which was estimated by a kinetic study. A correlation was found between partial pressure dependences and the K/Cu ratio caused by sublimation of copper, but it was slight. On the other hand, the activation energy of the deactivated catalyst tended to increase significantly with the increment of the K/Cu ratio.
Furthermore such an increase tendency of activation energy was utilized for predicting catalyst life. As a result, the simulation applying the increasing tendency of activation energy showed a good agreement with the measured value after 2 years.
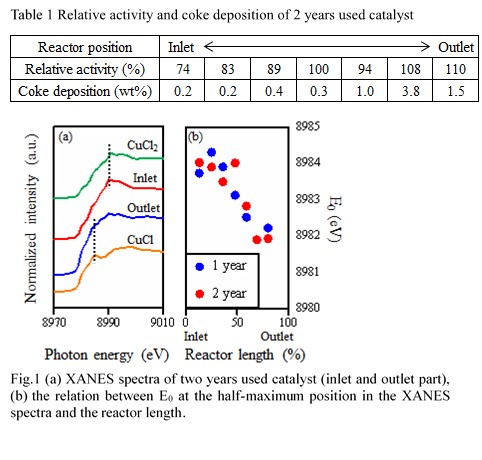
Oxygen-based oxychlorination is one of the efficient processes for producing 1,2-dichloroethane of a key material in vinyl chloride production. Despite abundant researches about the commercialized CuCl2-KCl/Al2O3 catalyst, its deactivation characteristics has not been fully understood. Besides, a carbon deposition property has not been examined in detail. Therefore, this study focuses on the clarification of the deactivation factor by XANES analysis of the commercialized catalyst as well as a measurement of the deposited carbon by temperature programmed oxidation (TPO).
The CuCl2-KCl/γ-Al2O3 catalyst was extracted from the industrial reactor after one- or two-years operation. The performance of the used catalyst was then evaluated by a fixed-bed reactor at laboratory scale. The deposited carbon was measured by TPO and the Cu oxidation state was investigated by Cu K-edge XANES analysis.
Table 1 shows a relative activity and the amount of coke on the two-years used catalsyt. The relative activity was defined as the conversion of the used catalyst devided by the conversion of the fresh catalyst. Althogh the catalyst at the inlet part was deactivated to 74-89%, the activity at the outlet part was still high. As for the amount of the coke, the more coke was observed at the outlet part, comparing to that amount at the inlet part. Fig. 1(a) shows XANES spectra of two years used catalyst. While the spectrum of the catalyst at the inlet part was similar to that of CuCl2, the catalyst at the outlet part was similar to that of CuCl. The Cu species of the outlet part catalyst were reduced state. Fig. 1(b) exhibits the relation between Edge jump energy (E0) and the reactor length. The E0 value was decreased from the inlet side to outlet side. The difference of the oxidation state might cause a deactivation of the catalyst and coke deposition property.
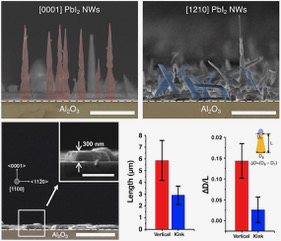
Organic-inorganic hybrid perovskites, such as methylammonium lead iodide (CH3NH3PbI3) have shown outstanding optoelectronic properties, promising extensive application in solar harvesting. Although most hybrid perovskite materials have been synthesized under solution process, recent demonstrations of the vapor phase synthesis of the hybrid perovskite nanostructures suggest there is an emerging interest in controlling their properties under highly-confined structures. In particular, single-crystalline 1D lead halide nanowires can be prepared via the vapor-liquid-solid (VLS) growth mechanism through the self-catalyzed growth mechanism, which then converted to hybrid perovskite. Since the conversion occurs on the preformed nanowire, electronic properties 1D perovskite depend on the original nanowire structure. In this study, we report the VLS growth of lead iodide (PbI2) nanowires on a c-sapphire (0001) substrate followed by conversion to CH3NH3PbI3 using methylammonium iodide, and confirm that the degree of perovskite conversion depends on the growth orientations of PbI2 nanowires. We observe two different growth directions; vertically-oriented [0001] nanowires and kinked nanowires. Photoluminescence (PL) measurements on each growth direction suggest that the oriented nanowires exhibit a higher degree of conversion compared to the [0001] oriented nanowires. In addition, [0001] oriented nanowires exhibit the position-dependent degree of conversion, depending on the presence of the catalyst tip on top of the nanowire. In particular, the conversion is observed both on the catalyst tip and the base of nanowire, suggesting methylammonium iodide incorporates into the nanowire either by vapor transport and surface diffusion. Our observation indicates that the vapor phase conversion of PbI2 to CH3NH3PbI3 is a diffusion-limited process. This finding is an important step towards a structural engineering of perovskite nanowires.
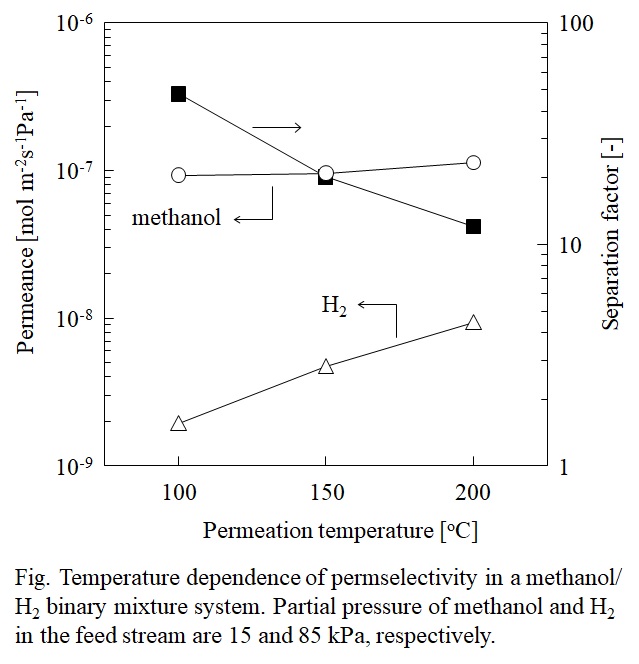
Methanol is an important industrial chemical and a raw material for the synthesis of various chemicals. It is mainly produced from synthesis gas, but the thermodynamic equilibrium in methanol synthesis results in a low conversion per pass. The use of a membrane reactor to improve the conversion per pass of the methanol synthesis has been considered. However, simultaneous membrane separation of methanol and H2O from H2 at high temperature is still difficult.
In this study, we developed a new concept on chemically stabilized ionic liquid (IL) membranes which are organosilica membranes with IL like properties prepared from silylated ILs. ILs have been used in membrane separation for more than a decade due to their special features such as non-volatility, thermal stability and ability to dissolve a large range of organic molecules. Gas/vapor permeation tests revealed that permeation separation performance of the membrane is strongly controlled by the affinity of the permeate molecules toward the IL, and its permeation and separation mechanisms are explained by a combination of the selective adsorption/pore blocking effect and the solution-diffusion mechanism.
The separation factors for methanol/H2 in the binary system were 12–48 in the temperature range from 100 to 200 °C. The membrane also showed high H2O/H2 separation performances in methanol/H2O/H2/CO2 quaternary system, and the presence of H2O and CO2 in the feed stream did not affect the methanol permeation performance. Methanol and H2O permeances of the developed membrane were higher than those of the Li-Nafion membrane, and comparable to values of the zeolite membrane. The silylated IL-derived organosilica membrane showed an excellent potential for the simultaneous separation of methanol and H2O from methanol synthesis gas.
Synchrotron X-ray powder diffraction has begun to stretch its strategic presence from the sheer determination of crystal structures into the mechanistic and kinetic information of microporous zeolite materials. Not only can this technique be used to reveal the internal framework structure of zeolites and their active sites, but it can also be combined with other analytical tools to suit the particular needs for in situ or operando gas storage and separation and catalytic studies.
With a keen interest in materials and catalysis, our research group has focussed on the fundamental aspects of zeolite materials. We have developed a new structural methodology by combining state-of-the-art synchrotron X-ray facilities with other characterisation techniques, that can:
(i) Directly visualise how organic molecules behave and interact with the BAS of zeolites.
(ii) Provide mechanistic information for important industrial processes.
(iii) Engineer new catalytic system for industrial processes.
(iv) Explain the reaction mechanisms and kinetics of new biomass conversion reactions.
(v) Design a novel enzyme-mimic inorganic system.
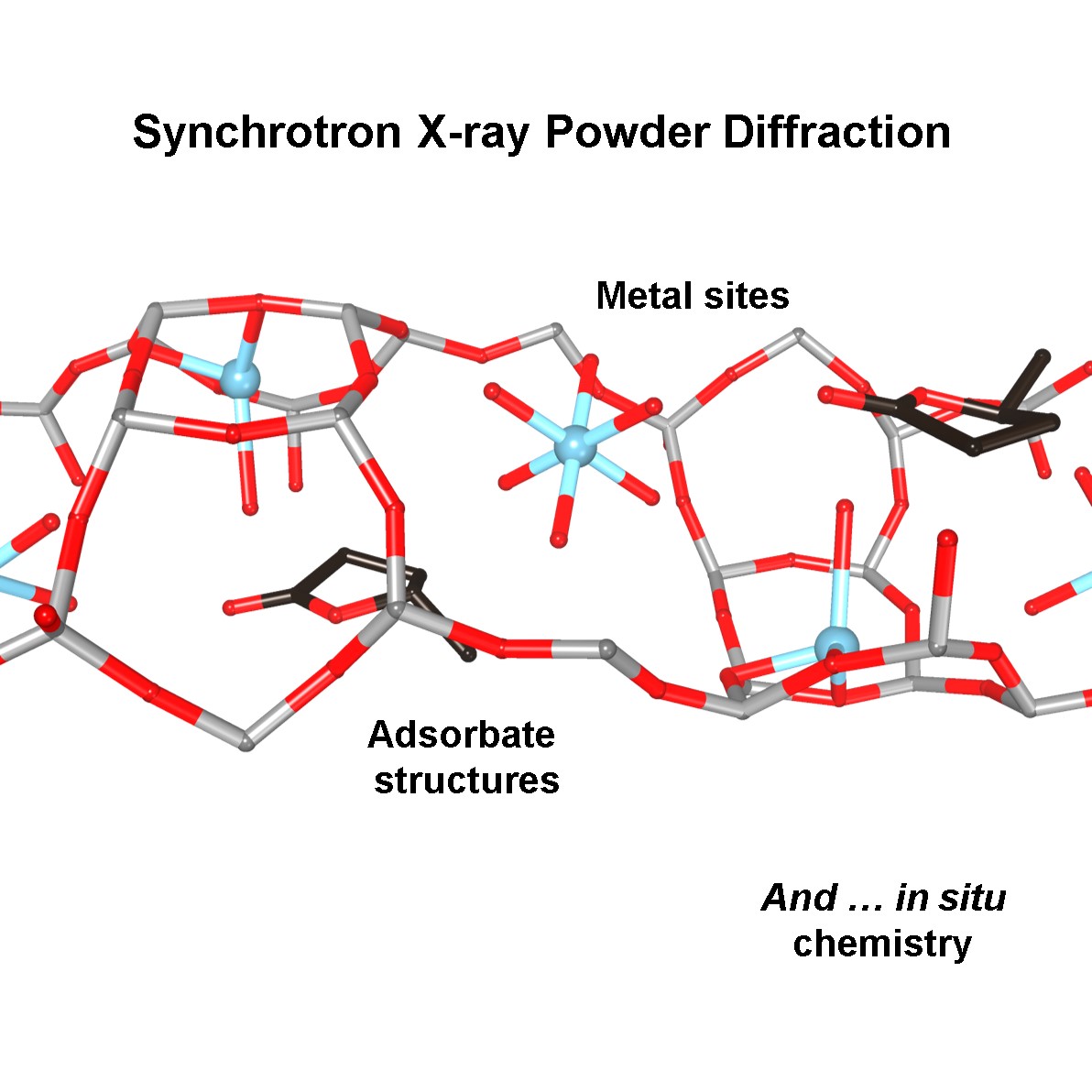
Atmospheric pressure plasma has been applied to induce a variety of unique reaction fields by supplying highly reactive species such as electrons, radicals, and ions. Among them, plasma reaction combined with water-based system has attracted intense attractions because plasma-water interaction generates various reactive species from dissociation of water molecule. Herein, applications of plasma-water interaction for fabrication of nanomaterials and treatment of waste water are introduced. Each application shows unique reaction system using various kind of plasmas and investigate the effect of plasma-water interaction. For fabrication of nanomaterials, plasma-assisted anodization and simultaneous reduction and immobilization of silver are described. Anodization of copper and zinc could be conducted using plasma in deionized water without addition of electrolyte. In this study, plasma-water interaction could increase electrical conductivity of deionized water by plasma electrolysis and trigger anodizing behavior. Meanwhile, silver nanoparticles were synthesized from reduction of silver nitrate with assist of plasma which provides reductive species. Especially, the reduction of silver could be accelerated by adding ethanol of which dissociation provides additional reductive species. When this process was conducted on the cellulose fiber, the reduced silver nanoparticles were simultaneously immobilized on the fiber. Treatment of waste water describes decomposition of phenol solution and immobilization of fluorine in fluorine-containing waste water. Decomposition of phenol aqueous solution was performed using in-liquid arc. Both oxidation and thermal decomposition were considered as the main decomposition mechanism. The oxidation occurred by hydroxyl radicals obtained from dissociation of water while the thermal decomposition was due to high enthalpy transfer from the plasma arc. The immobilization of fluorine was conducted to capture fluorine in waste water containing high concentration of HF. Alumina synthesized in situ by plasma could capture fluorine and significantly decrease the concentration of fluorine ion.
High aspect ratio nano-particles arising from the exfoliation or fragmentation of layered materials are widely used as fillers of polymers. Ultrasound irradiation is one of the potential methods to obtain the fillers rapidly and effectively. To keep up with the increasing demand of the filler production, ultrasound apparatus is expected to be developed to a large scale production process. However, ultrasound apparatus has been built only on an empirical basis since characteristic of the ultrasound and mechanism of layered compound fragmentation has not been revealed yet. In this study, the dependence of ultrasound irradiation conditions on fragmentation rate of the layered material, α-zirconium phosphate (α-ZrP), was investigated to construct the design methodology of the ultrasound process. Ultrasound generated from a horn mounted on a transducer with frequency of 20 kHz was irradiated to the α-ZrP aqueous suspension containing an intercalation agent. The fragmentation rate was evaluated by measuring the time course change of light transmittance of the suspension. The fragmentation rate was formulated with the proprietary developed fragmentation rate equation. The concentration of the α-ZrP and the ultrasound power input were varied and the fragmentation rate constants could be obtained by the linear fitting of the rate equation. It was found that the rate constant was linearly increased to the amount of ultrasound power input per unit weight of α-ZrP. Next, 4 types of sonication vessels having different volume were used to examine the vessel size effect. The smaller vessel used, the higher rate constant was obtained, and the rate constant was inversely proportional to the one-half power of volume. Eventually the fragmentation rate model could predict experimental results successfully, and the reactor model with the rate equation showed that a loop-type ultrasound reactor system with the smaller sonication vessel was promising for intensification of the ultrasound process.
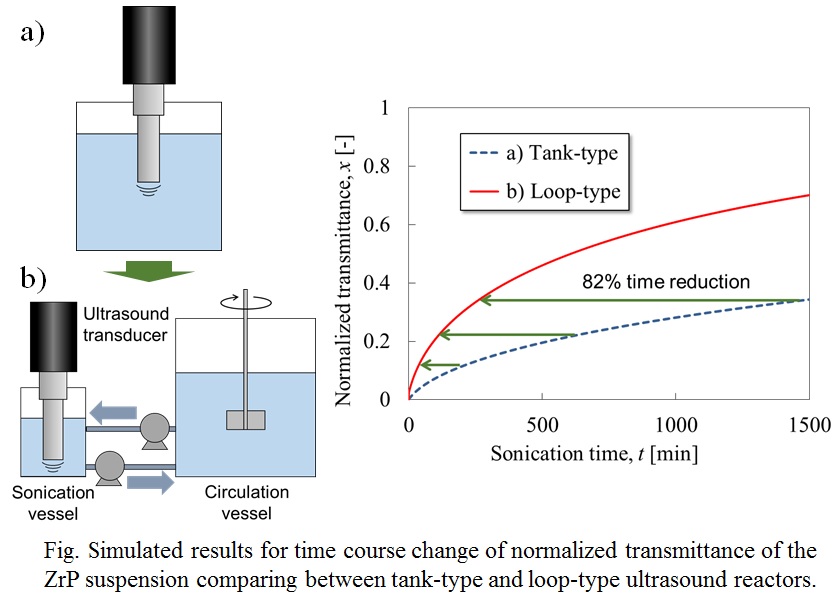
Microreactor technology as a promising tool of process intensification has received many research interests in the field of chemical and process engineering over recent decades. In this respect, the use of slug flow processing in microreactors represents an attractive alternative to carry out operations in biphasic systems (e.g., liquid-liquid extraction, gas-liquid/liquid-liquid reaction with or without the presence of homogeneous catalysts). The improved system performance can usually result from a precise manipulation of slug flow (e.g., uniform bubble/droplet dispersion, narrowed residence time distribution), and its superior mass transfer properties (e.g., high interfacial area, good mixing due to strong internal circulation). Up to now, a deep and fundamental understanding into mass transfer in slug flow microreactors is still limited, rendering the interpretation of the involved microreactor system somewhat empirical. When it comes to the emerging application areas, a number of opportunities exist for the use of slug flow microreactors within the context of green and sustainable chemistry, especially related to biomass conversion. In this presentation, our recent research highlights on mass transfer characterization in slug flow microreactors will be firstly summarized. The liquid-side mass transfer coefficient can well described based on the penetration theory, provided that the additional contribution of internal circulation within the droplet and slug under various operating conditions is properly considered. Then, the promising applications of slug flow microreactors in biobased chemical processes will be discussed. Typical examples include liquid-liquid reactive extraction of lactic acid, the synthesis of furanics (i.e. 5-hydroxymethylfurfural and furfural) from C5/C6 sugars in a biphasic system, the aerobic oxidation of 5-hydroxymethylfurfural to promising polymer building blocks (e.g., 2,5-diformylfuran), and enzymatic biodiesel synthesis. The microreactor operating principles, the slug flow mass transfer characteristics, the intensification potential as well as the (catalytic) chemistry insights will be addressed particularly.
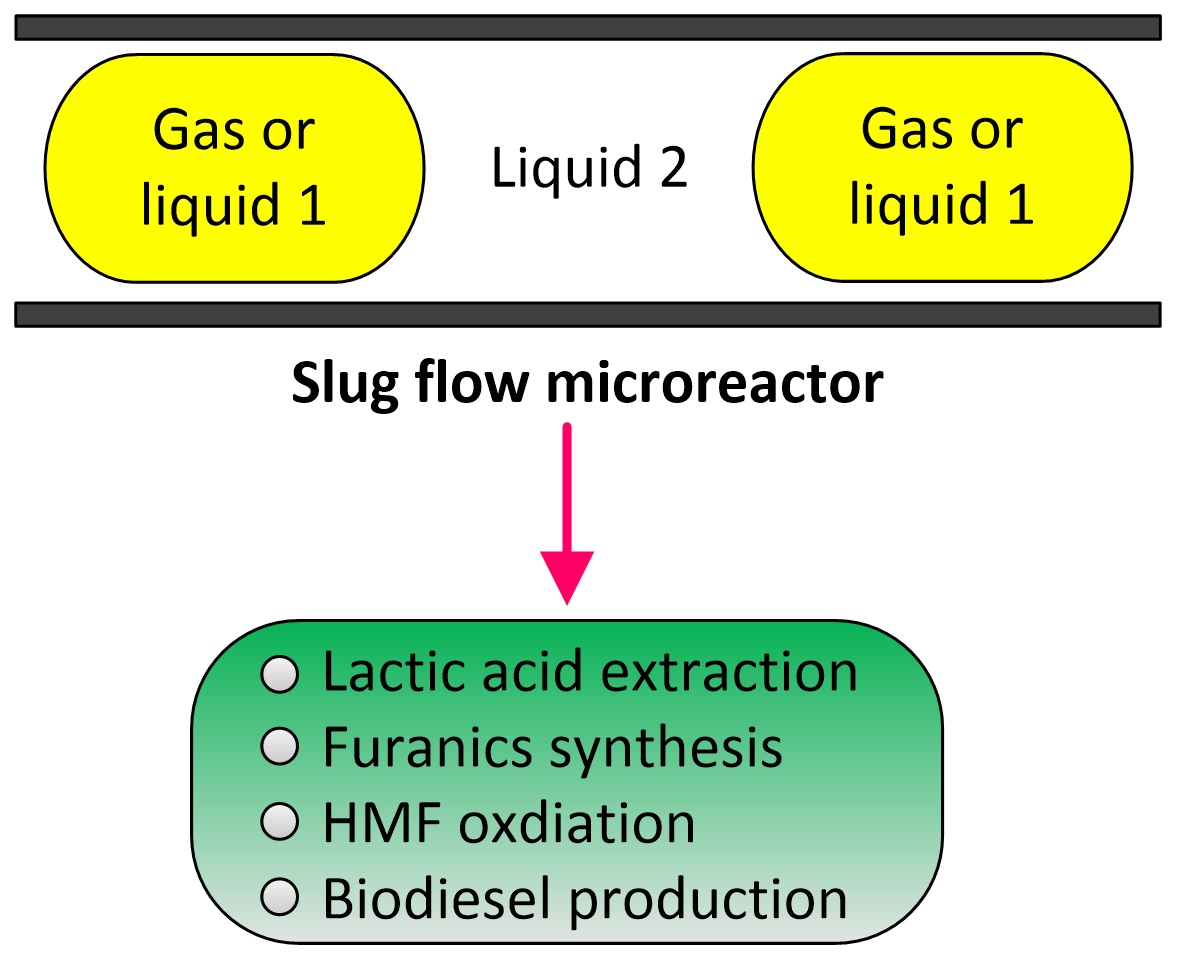
Recently falling film microreactors (FFMRs) are getting popularity for fast and exothermic gas-liquid reactions because of large specific surface area. Although FFMRs attain excellent mass transfer between phases, chemical reactions are often limited by diffusive mass transport in liquid phase. Therefore, this study aims to design novel reaction plates for promoting passive mixing and to derive the further potentials of FFMRs. First, micro-baffled plates with 2-dimensional structure are investigated numerically and experimentally in a systematic manner. The results indicate that the 2-dimensionally-structured baffles are not effective to enhance the overall mass transfer, although they can drastically change the wavy shape of gas-liquid interface and thicken the liquid film. A CFD model involving gas-liquid reaction is newly developed for the analysis of 2-dimensional wavy falling film. The CFD simulations show that gas absorption rate increases drastically in thin liquid film around the baffles while it decreases in the liquid film thickened between the baffles, and hence, the overall gas absorption rate slightly decreases compared to the standard plate without baffles. Subsequently, the micro-baffled plate with 3-dimensional structure is designed and examined. Fig. 1 shows the results of CFD simulations assuming that flat liquid film is formed on (a) 2-dimensionally-micro-baffled plate and (b) 3-dimensionally-micro-baffled plate. The vector plots in the figure show that the slantwise micro-baffled plate promotes secondary flow around the baffles, which is not observed on the 2-dimensionally-micro-baffled plate. This secondary flow is expected to enhance mass transfer. A CFD model being developed to resolve mass transfer across 3-dimensionally-wavy falling film will examine the effect of plate design and flowrate on gas absorption rate.
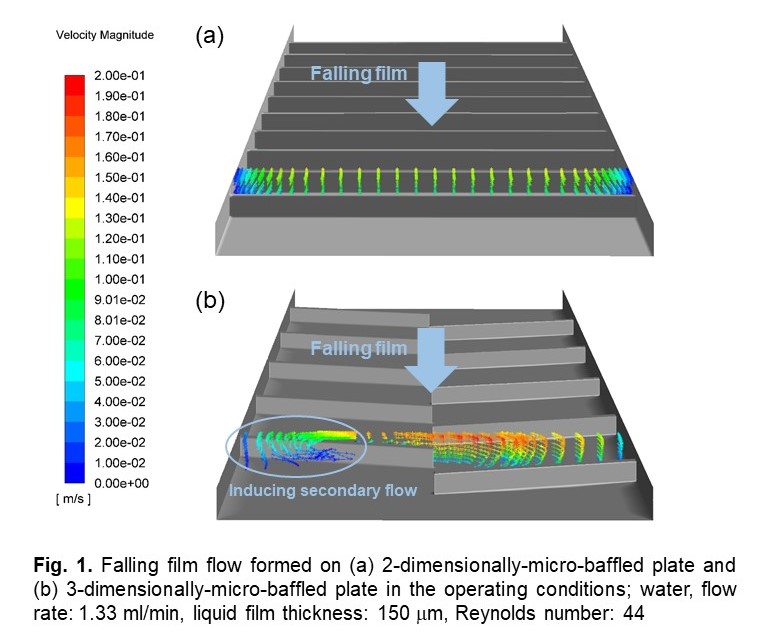
In a falling film microchannel, the interphase mass transfer strongly depends on the liquid-phase mass transfer behavior. The flow pattern of liquid film is predominantly laminar. And the liquid-side mass transfer relies heavily on molecular diffusion. Effectively improving the liquid film flow characteristics is the fundamental way to enhance the interphase mass transfer in falling film microchannels.
In this project, micro-mixing zones and wavy channel are configured respectively to improve the flow and mass transfer behavior of the liquid film in the falling film microchannel. The intensification mechanismes are systematically studied by experimental research and numerical simulation.
The results show that the fluid flow and mixing are improved by staggered flow mode in the double-side falling film microchannels with micro-mixing zones. Three flow patterns are observed inside the liquid film, including direct flow, cross flow, and vortex flow when the falling liquid film crosses the micro-mixing zones. Both cross flow and vortex flow intensify the surface renewal and turbulence of the liquid film, which is the fundamental reason for the enhancement of the liquid side mass transfer. The liquid side mass transfer coefficient of CO2 absorption into water is increased 7–14% by the configuration of micro-mixing zones. As the liquid flows on the falling film wavy microchannel, the liquid film resonates with the wavy microchannel wall. The surface fluctuation of liquid film can increase the gas-liquid interface area, and eddy might perturb the bottom of liquid film to enhance the mass transfer. Compared with flat microchannel, the falling film wavy microchannel increases the efficiency of CO2 absorption into water by 41%. The results suggest that falling film micorreactor with micro-mixing structrues can effectively enhance the interphase mass transfer behavior.
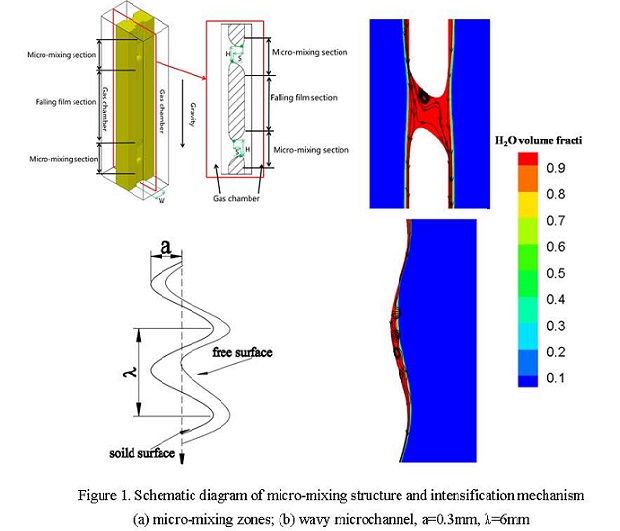
A large capacity micro-channel reactor called the Stacked Multi Channel Reactor (SMCR) was developed. SMCR enables having a larger capacity up to several tens liters by using a concept of numbering up of the channel than conventional micro-channel reactors and applying advantages of micro-channel reactor such as rapid heat and mass transfer to industrial chemical processes. Conventional micro-channel reactors have two dimensional structural junction such as Y-shaped and T-shaped for fluids mixing. These structures are not suitable to arrange numerous channels efficiently in an apparatus. Three dimensional structural junction for fluids mixing was adopted in SMCR. The SMCR consists of plates which have multi-channels on both sides and through-holes which connect the channels on both sides of the plate three dimensionally. This structure enables efficient arrangement of numerous channels on the plate and increasing the number of channels per unit volume of an apparatus by stacking the plates. In this work, the applications of SMCR to multi-stage continuous process, such as extraction, absorption and reaction, were investigated. Extraction experiments were carried out by using single channel equipment and SMCR bench scale equipment. The bench scale equipment consists of 3 SMCR and each SMCR has 24 channels with an inner radius of 1 mm. Total processing capacity of the bench scale equipment is approximately 0.6 liters and the processing flow rate could be on the order of several tens kilograms per hour. The bench scale equipment successfully reproduced the extraction performance of the single channel equipment. In this result, it was confirmed that SMCR can increase the throughput by numbering-up method maintaining performance in a single channel. Other applications on absorption and reaction process will be also presented on the day of presentation.
Recently, catalytic wall plate milireactor (CWPMR) has been proposed for process intensification of highly exo- and endothermic reaction by taking advantange of its high catalyst loading, scalability, and relatively low pressure loss. Although numerical study is indispensable to optimize device dimensions and operating conditions, neither of present options, namely the most accurate particle-resolved and inexpensive lumped continuum approaches, are able to achieve afforable computational cost and sufficient accuracy, respectively. In this study, computationally-inexpensive, detailed porosity profile-based continuum model has been developed and utilized to investigate different performance indices of CWPMR condcuting methane dry reforming. The new porosity profile model for CWPMR was developed by using DEM simulations. The examined range of packed layer thickness to particle size ratio (Np) was 1 to 5. The simulation results were compared to the experimental results for validation. The simulation results showed that mass and heat transport properties and their associated reaction performance were strongly influenced by the porosity profile. The highest reaction rates occured in the top region of packed layer where the momentum penetration was also observed, while the reaction rates were negligibly small in other regions as shown Figure 1. Such information can help to understand the interaction of reaction and transport phenomena in the CWPMR, as well as to improve the dimensions and design of the reactor.
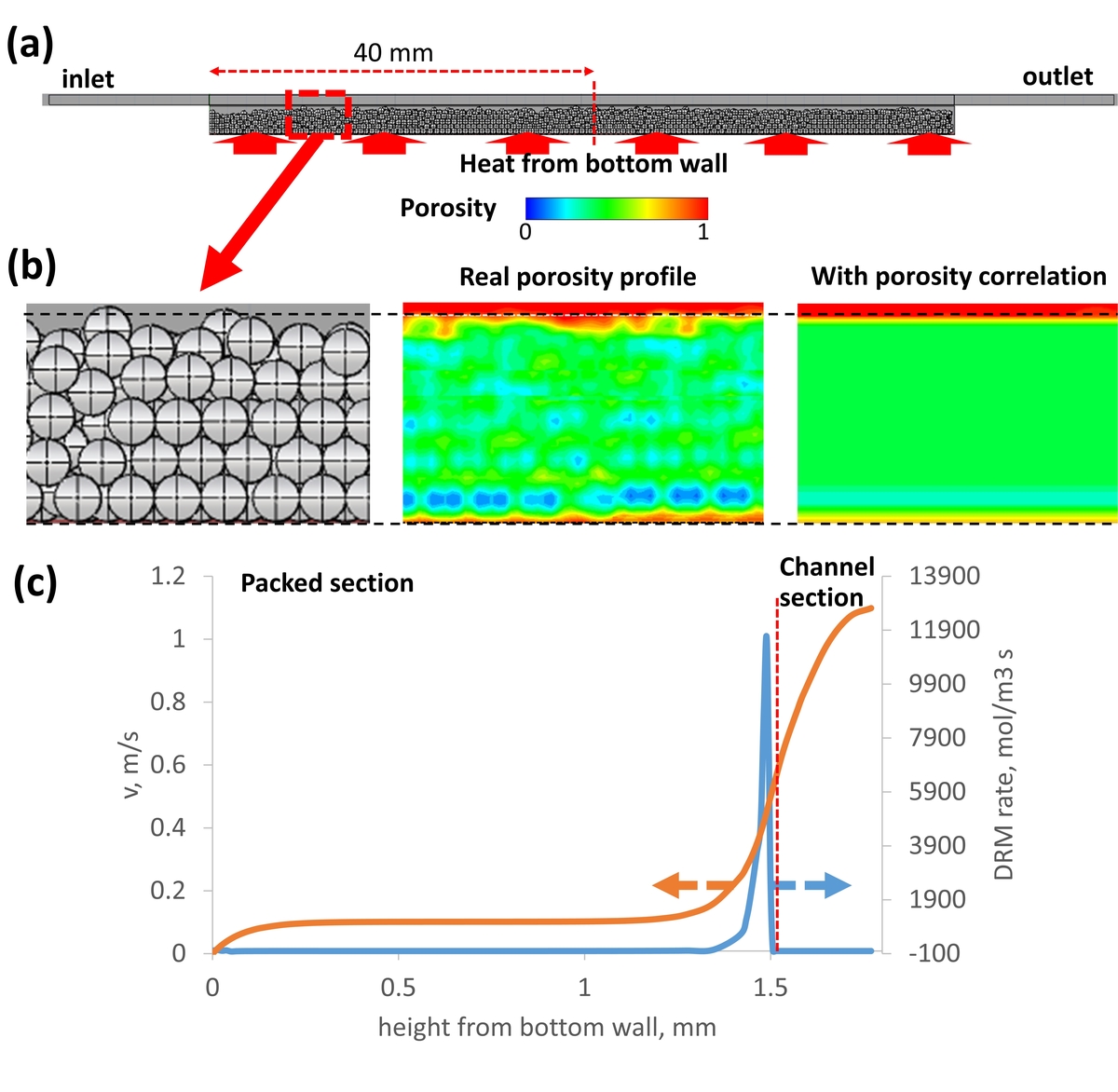
Most of our ‘playing field’ is in liquids where chemical reactions and separations occur. For enhancing the production rate of the target substance, various methods have been developed and applied. The rate was frequently controlled by mass-transfer or accurate triggering of reactions. Ultrasound has a power of thinning the liquid boundary film, enhancing mixing in narrow spaces and generating radicals with high accuracy. Also, ultrasonic irradiation is highly controllable and is easy to adapt to existing equipment. Works of ultrasound will facilitate reactions as well as separations. The irradiation ensures nucleation from supersaturated solutions. Solid surfaces of catalysts and silicon wafers are successfully cleaned. Reaction path appears only under sonicated conditions. Radical reactions are initiated by sonolysis of liquid medium.
Since ultrasound is pressure wave, the unique phenomena, cavitation occurs due to the pressure oscillation. Cavitation is characterized by generating fine bubbles in liquids followed by oscillation of those bubbles and eventual violent collapse to emit energies. Onset of cavitation dramatically enhances interfacial area; the field for reactions and mass-transfer are in play.
Ultrasonic irradiation to liquids causes a lot of physical and chemical changes, which are strongly related to our ‘games’ in chemical processing. Examples will be presented in the lecture to show a high potential for ultrasound as a game changer.

Poly(N-isopropylacrylamide) (PNIPAM) is a thermoresponsive polymer with a lower critical solution temperature (LCST). Poly(2-hydroxyethyl methacrylate) (PHEMA) has a high mechanical strength, so that a copolymer of poly(NIPAM-co-HEMA) is expected to be applied as advanced functional polymeric materials in various fields, such as sensors, actuators, and drug delivery system.
When ultrasound is irradiated to a solution containing vinyl monomers, the radical species are generated due to thermal decomposition of the monomers and then radical polymerization takes place without any chemical initiator. The synthesized polymer decomposes due to the high shear stress. Therefore, the polymer with a narrow molecular weight distribution can be obtained. It is expected to control the properties of the copolymer by controlling the condition of ultrasonic irradiation.
In this research, poly(NIPAM-co-HEMA) was synthesized using ultrasonic irradiation in a mixed solvent of water and ethanol without any chemical initiator. The effects of the ultrasonic power intensity, the ultrasonic irradiation time and the monomer molar ratio on the responsive temperature of synthesized polymers were investigated.
The responsive temperature of the copolymer obtained at the completion of the reaction linearly increased with the copolymer composition. The experimental results were in agreement with the estimated values by copolymer composition, which was formulated according to the results obtained by using chemical initiator (Shen et al., Colloid Polym. Sci., 2006, 284, 1001). However, the responsive temperature of the copolymer obtained in the early stage of the reaction was higher than that estimated by the copolymer composition. These results suggested that some parts of the polymer chain have higher NIPAM composition than overall composition because of the difference in the reactivity of the monomers. In conclusion, the composition and responsive temperature of the copolymer can be controlled by changing the ultrasonic irradiation time.
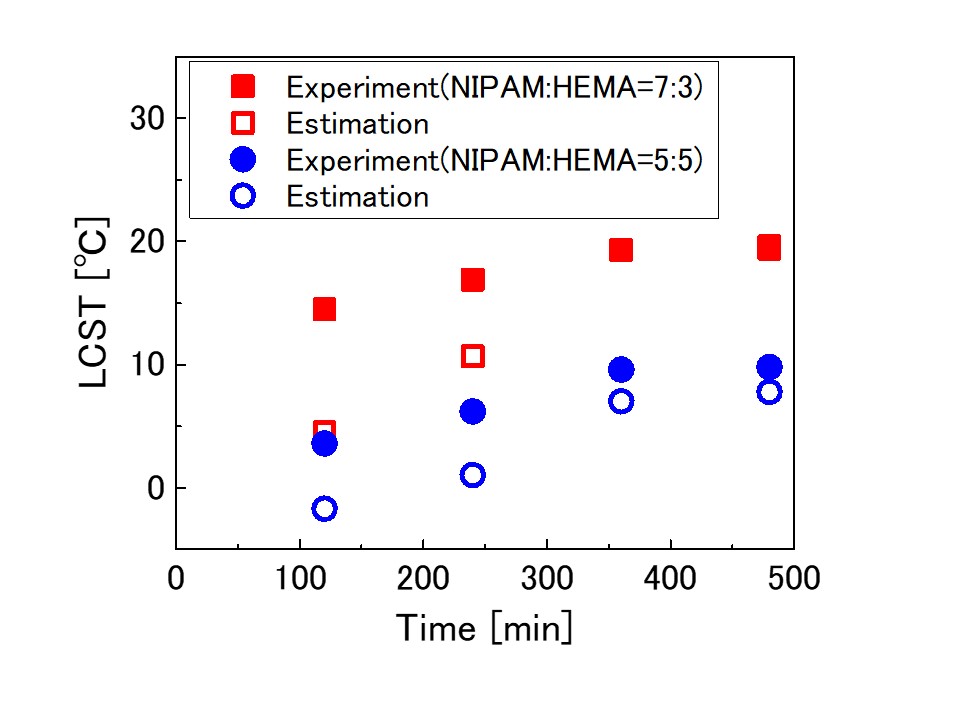
When ultrasound was irradiated to water, ultrafine bubbles were generated. Effects of ultrasonic power and frequency on ultrafine bubble concentration and diameter were investigated. Number concentration of ultrafine bubbles increased with time and approached asymptotically to an equilibrium value. Increase rate of ultrafine bubble concentration became higher as ultrasonic power increased. The equilibrium number concentration of ultrafine bubbles increased with decreasing frequency. The mode diameter of ultrafine bubbles was within 90 -100 nm. Effects of ultrasonic frequency and irradiation time on the mode diameter was small.
On the other hand, when ultrasound was irradiated to water containing high concentration of ultrafine bubbles (ultrafine bubble water), number concentration of ultrafine bubbles decreased with time and approached asymptotically to the equilibrium value. Reduction rate of ultrafine bubble concentration became higher as ultrasonic power decreased.
Generation and elimination of ultrafine bubbles by ultrasound were modeled to analyze experimental data. The generation rate coefficient of ultrafine bubbles increased with decreasing frequency because cavitation collapse became stronger. The reduction rate constant of ultrafine bubbles at 488 kHz and 1 MHz in ultrafine bubble water became much higher than those in ultrapure water due to aggregation and floatation of ultrafine bubbles. The equilibrium number concentration increased with decreasing frequency as shown in Figure 1. The results calculated by our model were in good agreement with the experimental data. The ultrafine bubble generator or eliminator using ultrasound is compact, simple in operation and produce or eliminate ultrafine bubbles in a short time. This method is contamination-free because the liquid pump and mixer are unnecessary.
Figure 1 Change in number concentration of ultrafine bubbles by ultrasonic irradiation to ultrapure water and ultrafine bubble water for different ultrasonic frequency at 15 W of ultrasonic power.
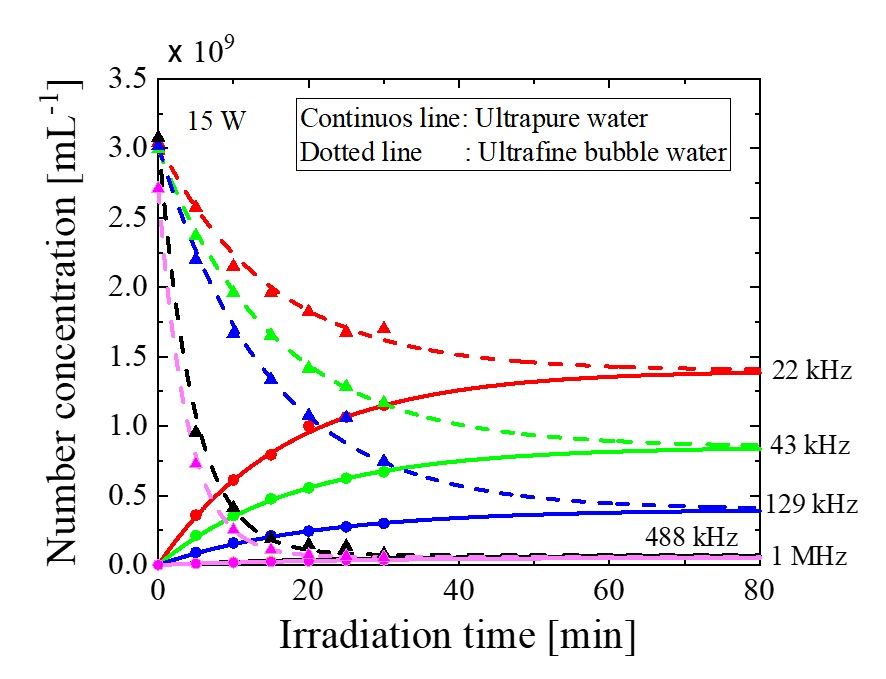
CO2 methanation (CO2 + 4H2 → CH4 + 2H2O, ΔH0298K = -165 kJ/mol) is one of the promising routes for converting CO2 into a useful resource of CH4. In a previous study, we developed a spiral-type Ni/CeO2 structured catalyst for enhancing a catalytic performance by promoting mass transfer diffusion. The developed spiral-type structured catalyst showed a high processing capability for CO2 methanation. This study focuses on the addition of O2 in the methanation feed gas, expecting for direct heat supply to the reaction field by H2 combustion. Therefore, in this study, the methanation performance of the spiral-type Ni/CeO2 structured catalyst was evaluated with changed O2 concentrations in the feed gas in the range from 0% to 10%.
The structured catalyst was prepared by an impregnation method and a wash-coating method. The catalyst component of 75 mg was coated on each spiral substrate. The methanation performance of the catalyst was evaluated by plug-flow type reactor. Four spiral catalysts were set in reactor tube. The reaction gas of 400 ml/min (CO2: 10vol %, H2: 40-60 vol%, O2: 0-10 vol%, N2: balance) was fed to the reactor.
Fig.1 shows the effects of O2 concentrations on CO2 conversions over the spiral-type Ni/CeO2 structured catalyst. In the CO2 methanation system without O2, the spiral-type structured catalyst showed a high activity at relatively low temperatures. By the addition of O2 in the methanation feed, the catalyst displayed a high performance at lower setting temperature. The increase in O2 concentration had a remarkable effect on the improvement of the catalytic performance. In case of 10% O2 in the methanation feed, 57% of CO2 conversion was obtained at room temperature without external heating. The result indicated that the generated high heat energy due to H2 combustion accelerated CO2 methanation on the spiral-type structured catalyst.
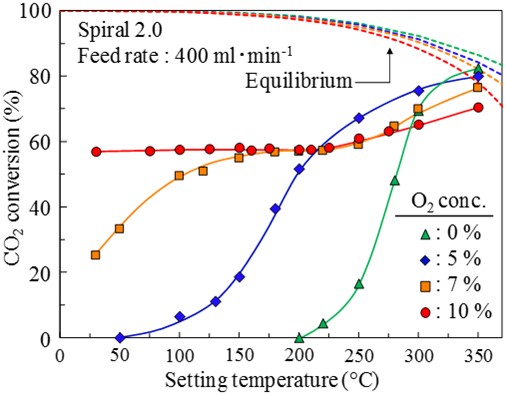
Nitridation on mesoporous silica by ammonia gas forms framework substituted nitrogen, and it has been known to work as base catalytic sites. Since the first attempt to use them as catalysts, several reactions have been catalyzed and further functionalization broadened applicable reactions. However, the detailed quantitative analysis of each silanamine site and their contribution toward catalytic reaction have been insufficient, and further study has been required for the future design of the catalyst
In this study, a series of nitrided mesoporous silica SBA-15 samples was synthesized at different nitridation temperatures. Quantitative analysis of formed silanamine species was conducted by both bulk and surface analyses (i.e., chemical analysis and XPS, respectively). For the quantitative analysis of each silanamine species, we used narrow scan spectra of XPS. The careful deconvolution of the spectra of N 1s level revealed the sequential formation of primary to tertiary silanamine species. The prepared samples were applied to several Knoevenagel and aldol condensation reactions as catalysts. The contribution of each silanamine species to the reaction was deconvolved by mathematical approach. Based on the result, the catalytic activity was in the order of primary > secondary ~ tertiary silanamine species. The order was different from the general textbook-order of the basicity of amine. This would be explained by more influential steric effect due to the formation of secondary and tertiary silanamine site inside the mesopore wall. Furthermore, primary silanamine showed significantly high activity for the reaction involving aldehydes, which would be explained by the formation of imine species. The formation of imine species was confirmed by FT-IR measurement.
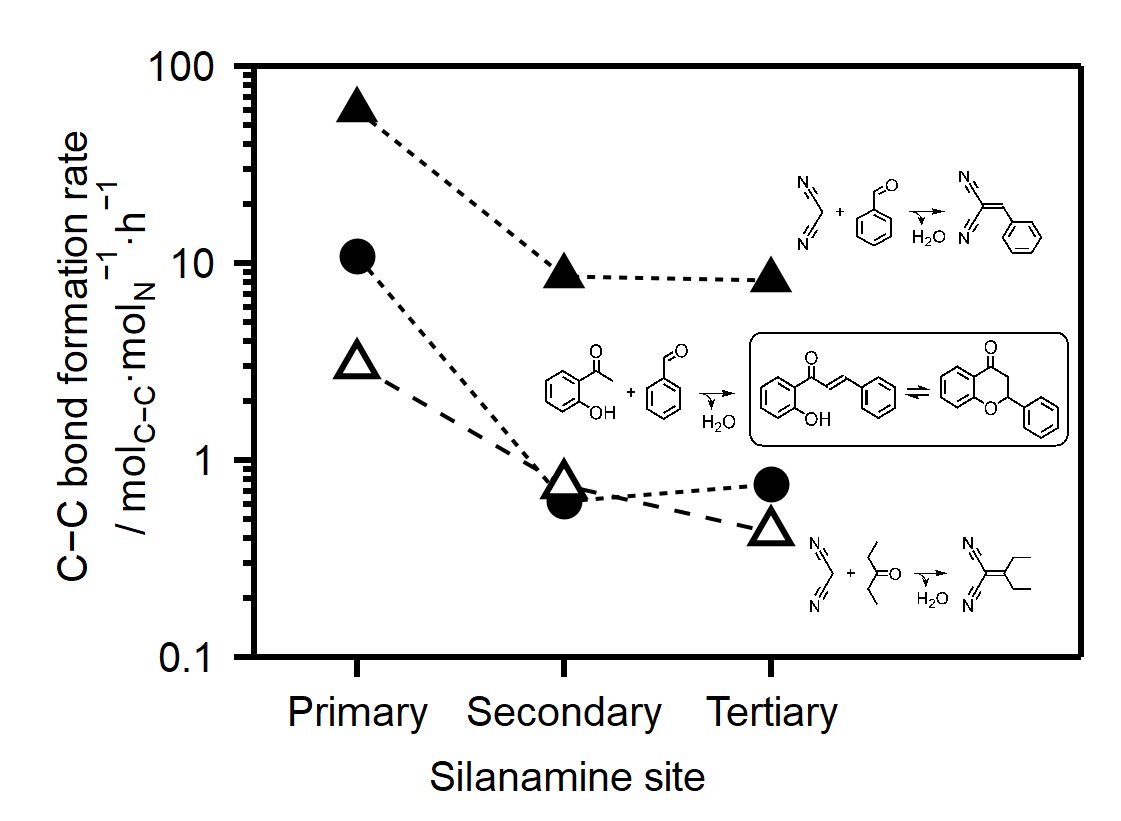
Mesoporous carbon that is a porous carbon material with regular meso-scale (2-50 nm) pores has large surface area and electrical conductivity, so it is used for various applications such as to catalyst support and for electrodes of electric double layer capacitors. Presently, mesoporous carbon is mainly produced by the template method. By the template method, however, it is difficult to form pores of over 20 nm, and the production cost is high due to the complexity of the process. In this study we have developed a method to produce mesoporous carbon by solid-solid reaction between carbon and iron oxide nanoparticles in dilute oxygen atmosphere. In the proposed method, the pore size of the mesoporous carbon can be arbitrarily controled by changing the size of the metal catalyst particles, and it may be advantageous in terms of time and cost for production because the process is simpler than the template method. Three composites were prepared by supporting iron oxide particles of 20-40 nm, 8-10 nm, and 4-6 nm respectively on carbon derived from coal tar pitch, and pore formation was conducted by heating each composite at 490 °C in the presence of oxygen. Nitrogen adsorption measurement of the prepared samples showed that pores of larger size were formed as the size of iron oxide particles became larger, suggesting the possibility of manipulating the pore diameter by selecting the size of iron oxide particles. In addition, by using iron oxide with a particle size of 10 nm or less, we succeeded in producing mesoporous carbon having pores of 7 to 50 nm by the proposed method.
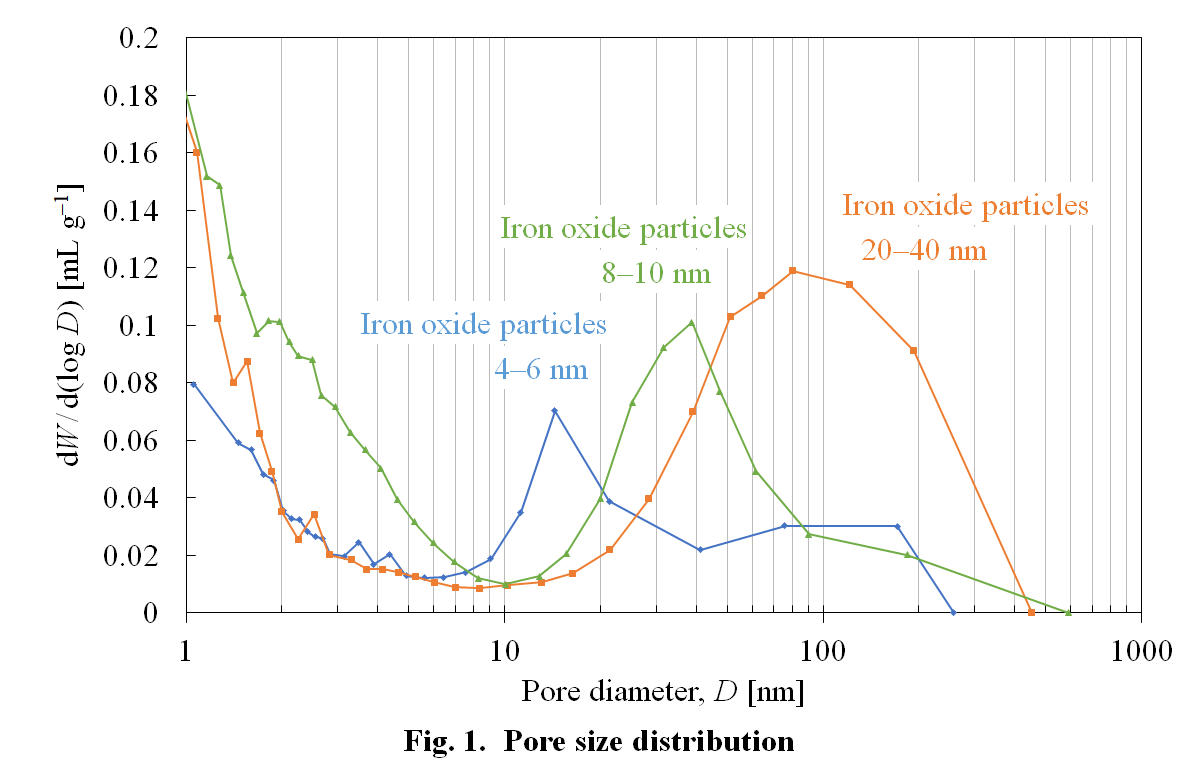
Nitrogen-doped carbons have attracted significant attention as catalysts for the oxygen reduction reaction (ORR) for fuel cell applications and H2O2 synthesis. Many research works have been devoted to the identification of the type of active nitrogen species doped in carbon framework to improve catalyst performance. For example, Nakamura et al.1 synthesized nitrogen-doped carbon catalysts possessing exclusively pyridinic or graphitic nitrogen species and have reported that pyridinic nitrogen present at graphene edge forms an active site. Our goal was to investigate the effects of the degree of crystallinity of carbon framework near active nitrogen sites on the ORR performance because it was anticipated to affect the binding strength of reactive oxygen species at active sites and potentially tune the selectivity to 2e– vs. 4e– reduction of O2. Tuning the degree of crystallinity of carbon materials may be accomplished through annealing in a conventional high-temperature furnace (>1200 °C). However, high-temperature annealing of nitrogen-doped carbons reduces their nitrogen concentrations. To minimize the loss of the nitrogen content of catalysts, high-temperature annealing may be conducted in a flow of NH3. However, it causes the decomposition of NH3, which generates radicals that gasify carbon materials. To address these challenges, we chose a microwave (MW)-assisted approach, which enables fast annealing of carbon materials.2,3 Here, we report that MW-assisted annealing of nitrogen-doped carbons in NH3 increases the degree of crystallinity while retaining porosity and active nitrogen species. We demonstrate that the annealed catalysts exhibit significantly higher ORR activity per pyridinic nitrogen site in an acidic electrolyte than a catalyst before MW-assisted annealing.
1. Guo, D., et al., Science 351, 361 (2016).
2. Mukai, S.R. et al., JP Patent 2015–23070; Kamatari, S. M.Eng. thesis (2015).
3. Ogino, I. et al., J. Energy Chem., 27, 1468 (2018).
Dehydrogenation reaction has attracted much attention for hydrogen production from hydrogen carrier such as methylcyclohexane and co-production of hydrogen and olefin or aromatic compounds from paraffin. Noble metals, such as Pt and Pd, are reported to exhibit high activity for dehydrogenation. Carbon is one of promising catalyst support because they have no acid/base sites, which causes undesired reaction such as cracking and isomerization. On the other hand, the metal fine particles on carbon support are easily aggregated during the catalyst preparation and the reaction, leading to deactivation of the catalyst, and Pt is active for C-C cleavage, causing low hydrogen selectivity. To maintain the metal particle size small, we employed a novel preparation method for carbon supported Pt fine particles by using an anion-exchange resin and Pt chloride anion as initial materials of carbon support and Pt, respectively. In addition, it has been reported that C-C cleavage reaction is suppressed by the addition of transition metal, such as Sn, Mn, and Fe, to Pt. Because Sn forms chloride anion in HCl solution, we considered that carbon supported Pt-Sn bimetal catalyst could be obtained by adding Sn chloride anion to Pt precursor solution in the preparation of Pt/C from the anion-exchange resin. In this study, the carbon supported Pt-Sn bimetalcatalysts with various metal compositions were prepared by using the anion-exchange resin and Pt and Sn chloride anions. Although Sn composition was smaller than the predetermined one, which was because Sn chloride melted and leached during the carbonization at 500 °C, Pt-Sn bimetal catalysts possessing 2-3 nm of metal particles were successfully obtained. Moreover, the metal loading of the obtained catalysts exceeded 10 wt% and hence they could exhibit high activity based on unit volume. We applied the prepared catalysts to dehydrogenation of paraffin to evaluate hydrogen production activity.
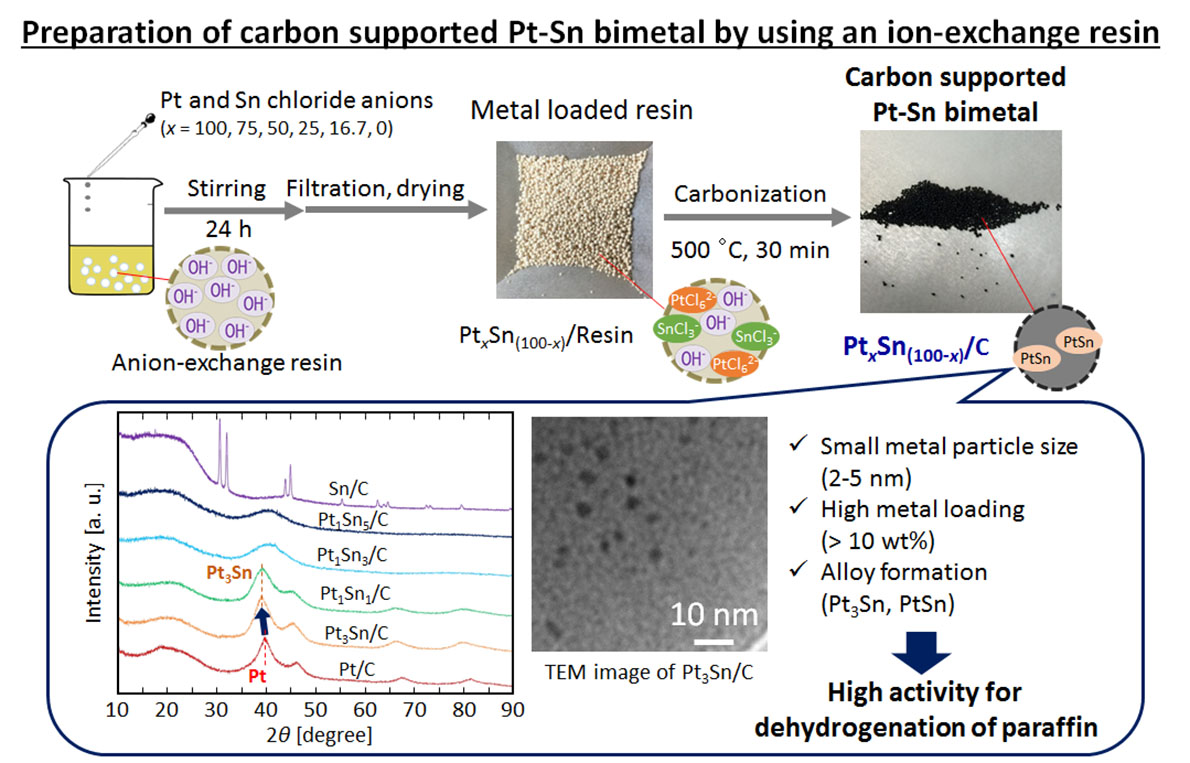
Polyoxymethylene dimethyl ether (POME) has gained much attention due to its unique properties such as high cetane number, better intermiscibility, and low soot and NOx formation. Therefore, POME has become a potential candidate for direct fuel or fuel additive in diesel engines. Conventionally, POME is produced through acetalization of formaldehyde with methanol by using acidic catalysts. Klankermayer and co-workers reported the homogeneous Ru and Co triphos-based catalysts with Lewis/Brønsted acidic co-catalysts for dimethoxy methane (DMM) or OME1 production from CO2 hydrogenation in methanol. To our best knowledge, POME synthesis from heterogeneous catalytic COx hydrogenation have not reported in the literature.
Wet impregnation method was used in current study for synthesis of X%Ru/BEA (X = 1, 3, 5) catalysts to produce POME from COx hydrogenation. POME synthesis experiments were conducted in autoclave batch reactors at 75 bar, 50 mL methanol and 100 rpm impeller speed. The prepared catalysts were characterized to check the catalyst structure, BET surface area and pore size distribution, Ru dispersion over BEA zeolite support and particle size, Ru reduction properties and metal-support interaction. Experimentally, 3%Ru/BEA revealed the best POME yield for COx hydrogenation at 125 °C under mentioned reaction condition. The optimized temperature range for CO2 hydrogenation over 3%Ru/BEA was reported as 150-175 °C. The adverse effects of water in product mixture and plausible reaction pathway were also investigated in presented study. The maximum OME1 and OME2 yields were 7.42 and 0.48 mmol/gcat.LMeOH, respectively at 150 °C, H2/CO2 ratio 3 after 2 h of reaction under same reaction conditions.
Isobutene is widely used as a starting material for alkyl t-butyl ether, isooctane, p-xylene, methyl methacrylate (MMA), butyl rubber and so on. Therefore, it is very important to establish an alternative route for isobutene production. Especially for us, it is preferable to obtain cheaply available isobutene for MMA production. Considering one of the routes, we have been studying the oxidative dehydrogenation (ODH) reaction of isobutane to isobutene as our aim. Our team has investigated various metal doped mesoporous silica catalysts and confirmed that SBA-15 modified with Cr and Mo (Cr, Mo-SBA-15) showed high performances for ODH reaction of isobutane. According to our investigation, the conversion of isobutane for SBA-15 and Mo-SBA-15 were quite low but Cr-SBA-15 showed the highest conversion of isobutane. However, in the case of Cr-SBA-15, it was quickly deactivated. On the other hand, in the case of Cr, Mo-SBA-15, the conversion of isobutene and the selectivity of isobutene were much higher than those of other catalysts. In addition, deactivation of the catalyst was remarkably suppressed than others. The binary metal system, Cr, Mo-SBA-15, had excellent functions for ODH reaction of isobutene.
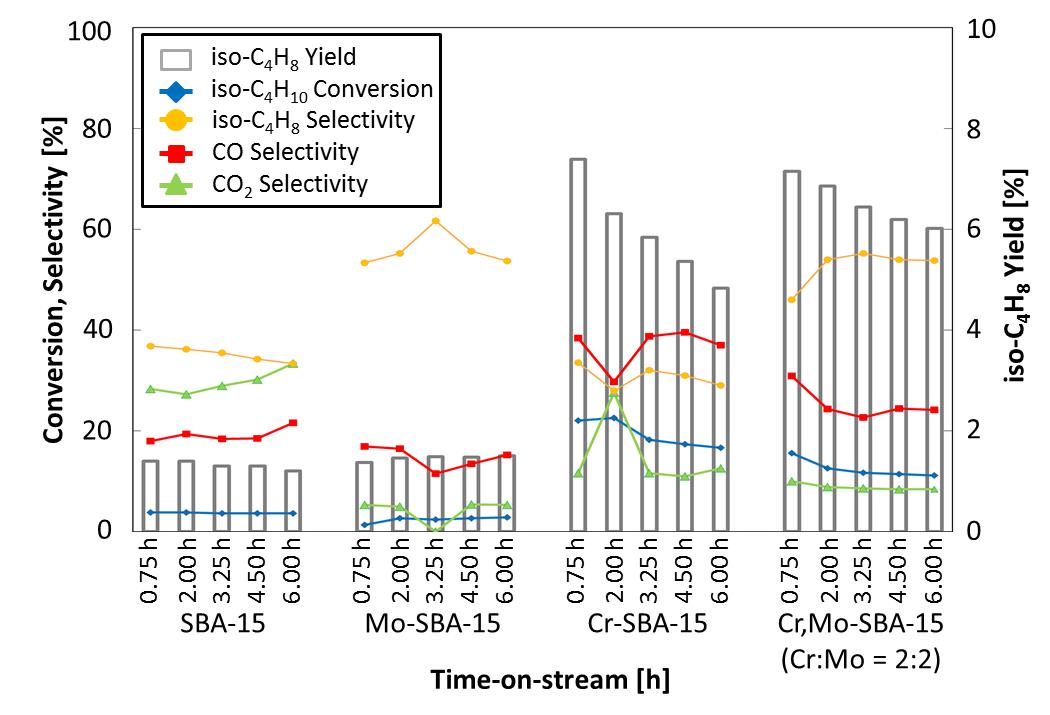
Propane dehydrogenation (PDH, C3H8 → C3H6 + H2) has gained much attention due to a direct production of valuable propylene. A metal sulfide is known to be a highly-active catalyst for PDH, however the loss of sulfide ion (S2-) from the surface frame structure by reduction with H2 leads to decrease of catalytic activity. In a previous study, we investigated a performance of SiO2 supported transition metal catalysts (Fe, Co, Ni, Mn, Cu) for PDH in the presence of H2S, expecting for replenishing S2- in the the surface frame structure of metal sulfide continuously. Among transition metal catalysts, a Fe/SiO2 catalyst showed a relatively stable performance with highly-active and selective for PDH with co-feeding H2S. In this study, we examined the effect of noble metal (Ru, Pd, Pt) addition to the Fe/SiO2 catalyst on its catalytic performance, in order to further enhance the dehydrogenation performance.
By the addition of noble metals, the catalytic activity of the Fe/SiO2 catalyst was largely enhanced. In particular, the Ru-added Fe/SiO2 catalyst showed a stable performance with high conversion and selectivity, comparing to that of the Pd- and Pt-added Fe/SiO2 catalysts. Figure shows the effect of the ratio of co-feeding H2S to propane (H2S/C3H8) on conversion over the Ru-added Fe/SiO2 catalyst. By increasing H2S/C3H8 ratio, a high conversion was obtained at the initial stage of the reaction, but the catalytic activity was decreased gradually. While, under low H2S/C3H8 condition, high stable performance was obtained. Almost no deterioration was observed for 3 h-dehydrogenation under 0.25 and 0.5 of H2S/C3H8. According to Fe K-edge XANES spectra, a high oxidation number of Fe species was confirmed. Such cation species could accept electron from intermediate species, which might promote the PDH performance.
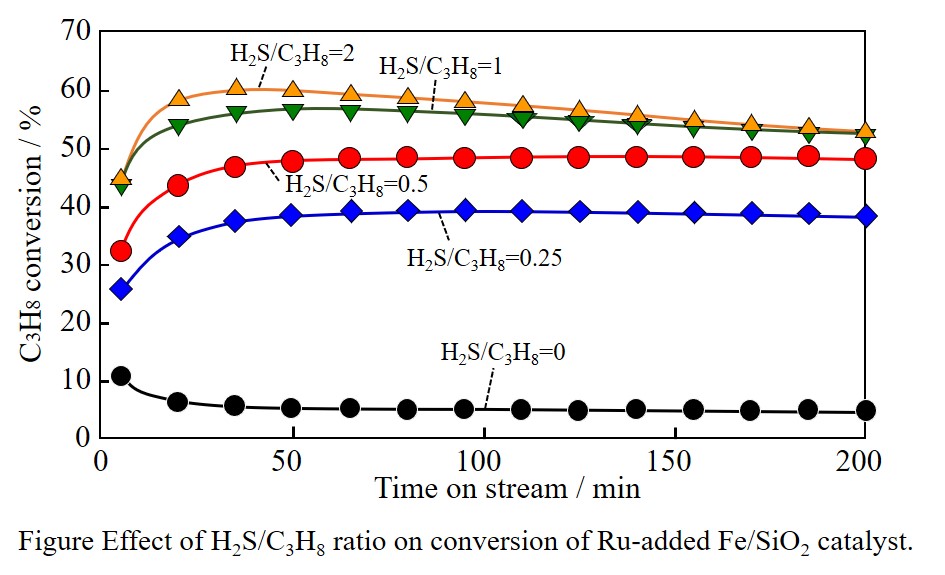
Biomass-derived polyols, such as glycerol, erythritol, and xylose, are attracting attention as renewable raw materials for basic chemical products, such as propanediols, butanediols, and 2-methyltetrahydrofuran. Because these polyols have high oxygen content (O/C = 1), deoxygenation reaction without C-C cleavage is required to convert them into the basic chemical products. In general, hydrodeoxygenation reaction is employed under pressurized hydrogen atmosphere and in the presence of metal catalyst containing Cu or Ru. To suppress oligomerization of polyols proceeding above 250 °C, catalyst with high activity at low temperature, that is, high metal dispersion, and without active site on catalysts support such as carbon support is needed. In conventional catalyst preparation methods including impregnation, it is difficult to achieve high dispersion on carbon support. To solve this problem, we focused on a new preparation method for carbon supported metal nanoparticle catalyst, which uses ion exchange resin as a precursor of carbon support. In this method, metal ions are loaded on an ion exchange resin by ion exchange and then the metal loaded resin is carbonized to convert the resin to carbon support. This study aimed at the preparation of carbon supported Cu catalyst by using the ion exchange resin as a precursor of carbon support and the investigation of hydrodeoxygenation reaction pathway of erythritol (C4 polyol) over the Cu/C catalyst. The Cu/C catalyst possessing 14 nm of Cu fine particles and 67 wt% of metal loading was successfully prepared by the ion exchange technique. We applied the obtained Cu/C catalyst to hydrodeoxygenation of erythritol to butanediols to investigate the reaction pathway by varying reaction time and hydrogen pressure. It was suggested that the main reaction pathway was 1,2-butanediol production from erythritol by sequential hydrodeoxygenation via 1,2,4-butanetriol.
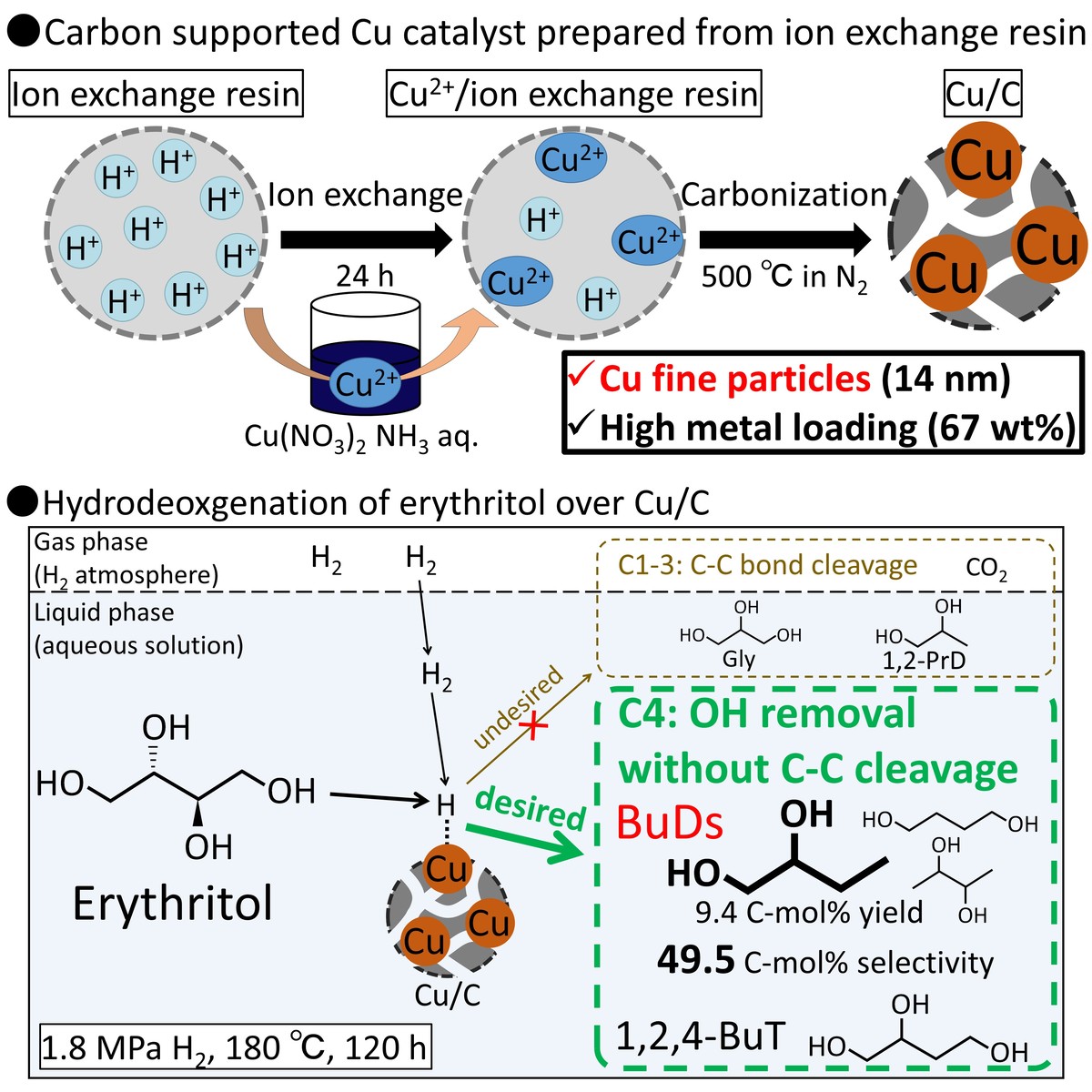
Alumina (γ-Al2O3) and silica(SiO2) have been widely used as adsorbents or catalysts/catalyst supports for hydrodeoxygenation in bio-oil, because of their high surface area, high thermal and chemical stability. The HDO is one of the promising methods for upgrading bio-oil by removal of oxygen-containing compounds. In this study, SiO2/γ-Al2O3 composite particles with different Al to Si ratios were synthesized by spray pyrolysis combined with sol-gel process. The spray pyrolysis method has advantages of synthesizing spherical micron-sized particles in one step with controlled size and morphology of the product particles. Also, mass production is possible with this method as it is a continuous reaction process. In order to control specific surface area and pore structures, the particles were prepared by adding CTAB as a template. The product particles were analyzed by BET, XRD, FE-SEM and TPD. Also, the produced gas and liquid products from HDO were analyzed by GC with TCD/FID.
This study reviewed the technologies that have been applied to hydrogen liquefaction processes so far. First of all, various types of hydrogen liquefaction processes were closely reviewed, and their advantages and disadvantages were compared. In particular, the effect of refrigerants and types of refrigeration cycles on energy efficiency of the process were analyzed. Then, recently developed innovative conceptual approaches to the hydrogen liquefaction process were reviewed. Lastly, a new approach was suggested to increase an energy efficiency of the hydrogen liquefaction process in South Korea.
Propane dehydrogenation (PDH) is one of the most promising candidates of propylene production process to meet the growing demand of propylene in the future. However, precious metals such as Pt have been required for high catalytic performance. Therefore, there is an urgent need to develop new catalysts based on non-precious metals for PDH reaction.
The scheme of PDH reaction is shown in Fig. 1. The initial reaction step from propane is dehydrogenation to propylene or cracking to ethylene and methane and then aromatic compounds can be produced through the excessive reaction. Therefore, it is required to increase the selectivity of dehydrogenation pathway and depress the excessive reaction. To meet these requirements, we focused on Brønsted acidity derived from Ga-species and diffusivity of zeolites which have large micropores.
As an ideal catalyst for PDH reaction, we have synthesized high silica Ga-Beta from dealuminated zeolite using dry gel conversion method. Ga species was well incorporated into *BEA structured zeolite frameworks and worked as Brønsted acid sites. The high silica Ga-Beta with Si/Ga ratio up to 177 can be synthesized. On the PDH reaction, synthesized Ga-Beta showed the highest propane conversion, the highest propylene yield and the highest selectivity of dehydrogenation on the initial reaction step in Ga-based zeolites. The combination of large microporosity and acid sites derived from Ga incorporated into the zeolite frameworks is likely to lead to the excellent catalytic performance of the Ga-Beta. Moreover, the Ga-Beta with higher Si/Ga ratio shows longer catalyst lifetime on PDH reaction since coke deposition rate was depressed by the decrease of the acid amount. This work provides new insights for PDH reaction over Ga based zeolite catalysts, which would contribute to the progress in activation and transformation of light alkenes to value-added chemicals.
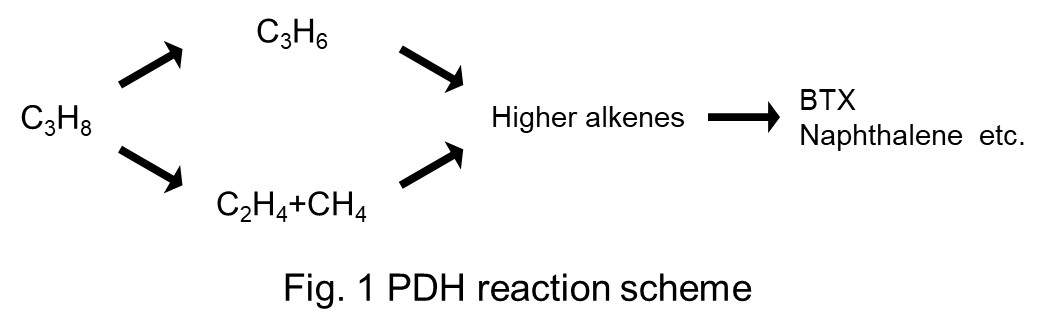
Formic acid is regarded as a promising energy carrier because hydrogen can be produced at mild conditions. Formic acid can be decomposed according to the following two pathways.
Dehydrogenation : HCOOH → H2 + CO2 (1)
Dehydration : HCOOH → H2O + CO (2)
Selectivity of the reaction pathway (1) is very important issue not only because low selectivity will lead to wasting much energy carrier, but also because CO is a poison for fuel cell catalysts in the subsequent conversion of hydrogen into electrical energy. Hence, an efficient catalyst for dehydrogenation of formic acid should be developed.
Considering the separation and reusability of catalysts, heterogeneous catalysts are more suitable for practical applications than homogeneous catalysts. Catalyst support is also crucial for H2 selectivity. Al2O3 or TiO2 support may have the activity to decompose formic acid without generating H2. In this work, carbon supported metal catalysts were used for formic acid decomposition.
Catalysts were prepared from a weak-acid-cation-exchange resin (WK11, Mitsubishi Chemical Corporation) and metal cation solutions. The resin and the aqueous solutions of [Pd(NH3)4]Cl2 and Ni(NO3)2 were stirred at room temperature for 24 h. Treated resins were filtered and dried followed by the heat treatment in N2 steam at 500 °C to prepare Pd/C, Ni/C and Pd-Ni/C catalysts.
Thermogravimetric analysis and powder X-ray diffraction patterns provided that the Pd and Ni particles were highly loaded and highly dispersed. A fixed bed reactor was used for the decomposition experiments in gas phase. Formic acid was able to be completely decomposed below 200 °C for all catalysts. H2 selectivity was above 85 % for Pd/C and Pd-Ni/C catalysts, indicating that they are suitable for H2 production from formic acid. Liquid phase decomposition was also performed.
2,5-furan dicarboxylic acid (FDCA) derived from the oxidation process of 5-hydroxymethylfurfural (5-HMF) is a valuable platform chemical for a wide range of industrial applications - particularly the bioplastic industries. This research aims to incorporate 1 wt.% palladium nanoparticles (Pd NPs) into vanadium(V) oxide (V2O5) which was used as an alternative heterogeneous catalyst for HMF oxidation. The incorporated Pd/V2O5 catalyst was prepared by colloid-chemical reduction step and immobilization step, where the combination of these two steps proceed differently - stepwise (ST) and simultaneous (SI) process. For the colloid-chemical reduction step, an aqueous solution of PdCl2.H2O (0.086 mmol Pd, 250 ml DI) was mixed with 2 wt.% of polyvinyl alcohol (PVA) solution - uses as a nanoparticle stabilizer blocking part of the active sites by covering the majority of the metal surface. The mixed solution, then, was reduced by adding slowly of 0.1 M NaBH4 solution. In case of ST process, adding 3 g V2O5 powder for Pd NPs immobilization was done after the reduction step while the simultaneous (SI) one was done at the same time. Morphology, particle size and distribution, as well as, the chemical composition of synthesized samples are characterized by various techniques i.e. TEM, BET, X-ray diffraction, UV-vis spectrophotometer. The modified Pd/V2O5 catalysts obtained from the two different processes (ST and SI) affected the surface characteristics – different Pd NPs size and distribution as well as specific surface area. The potential oxidation of 5-HMF over incorporated Pd/V2O5 catalyst was higher than the one without Pd NPs – raising 5-HMF conversion percentage about two times.
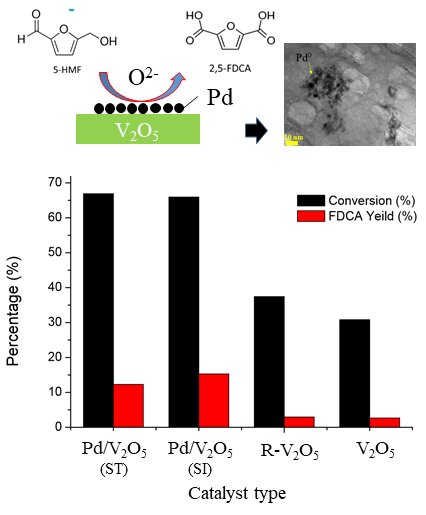
The allylic oxidation is one of important reaction in chemical industry to convert olefin into unsaturated aldehyde for valuable intermediates. It is generally reported that Bi-Mo oxide catalyst loaded on Co-Fe-Mo complex oxide support is effective for allylic oxidation. In this reaction, the oxygen atom in Bi-Mo oxide is consumed in allylic oxidation, gaseous oxygen is uptaken in Co-Fe-Mo complex oxide, and the lattice oxide of the support is transferred to Bi-Mo oxide to regenerate the active center. It is commonly reported that Fe atoms, which is dissolved in CoMoO4 matrix in the support, are responsible for the lattice oxygen transfer and this step is considered to be the rate-limiting in allylic oxidation. Therefore, the allylic oxidation activity can be enhanced by improving the dispersion of Fe atoms in the support. We herein investigated the effect of preparation condition on the dispersion of Fe atoms in the support and the catalytic activity for the allylic oxidation. Co-Fe-Mo complex oxide was prepared by co-precipitation method and the initial pH value of the metal precursor solution was varied to control the precipitation rate of each metal which can be related to the dispersion. When the starting pH value was high, the Fe atoms in the support was highly dispersed without forming greatly agglomerated Fe2(MoO4)3. Furthermore, the allylic oxidation of allylbenzene to cinnamaldehyde was performed by using α-Bi2(MoO4)3 loaded Co-Fe-Mo complex oxide prepared. The catalyst with higher dispersion of Fe showed the higher cinnamaldehyde yield, which could be due to the improvement of the oxygen transport capacity and the promotion of reoxidation of the active center.
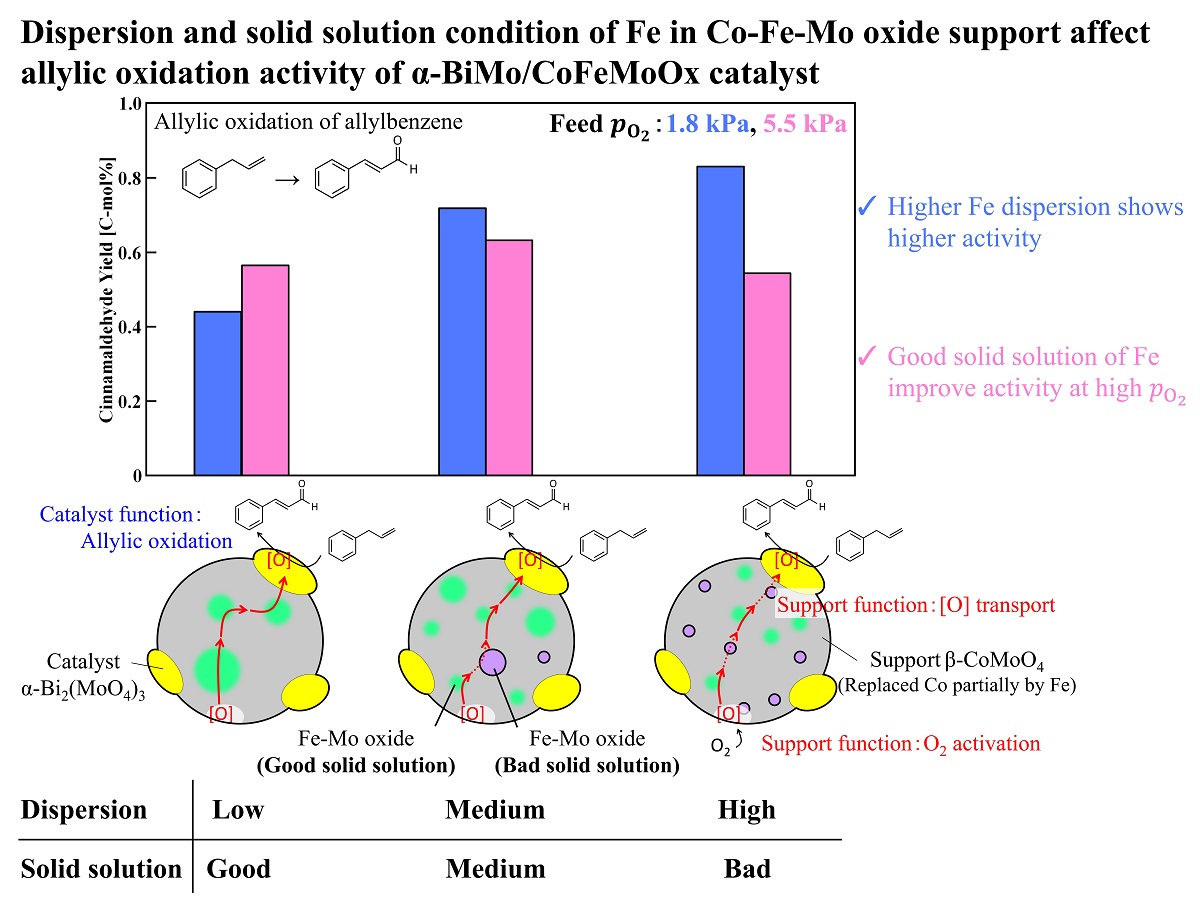
Benzene, toluene and xylene (BTX) are value-added chemicals as basic petrochemical products. These chemicals are mainly obtained from naphtha by the catalytic reforming process. Meanwhile, direct conversion of light alkanes to BTX is a promising process to utilize natural gas as a feedstock for their production. Zn2+-exchanged ZSM-5 (Zn-[Al]-MFI) zeolite was known to be an effective catalyst for converting ethane to BTX. However, the catalyst was rapidly deactivated by a coke deposition. Here, we examined incorporation of Ga into the zeolite framework (gallosilicate). Ga species in the zeolite framework is known to promote aromatization of light olefins. Additionally, gallosilicate zeolite has lower acidity compared to alminosilicate zeolite. In this study, we prepared Zn2+ ion exchanged MFI type galloaluminosilicate (Zn-[Ga, Al]-MFI) for ethane dehydroaromatization (EDA) reaction in order to improve catalyst life time as well as BTX yields.
Both Zn-[Ga, Al]-MFI and Zn-[Al]-MFI samples showed typical XRD patterns of MFI zeolite. From NH3-TPD spectra of the samples before Zn2+ exchange, the acidity is higher in the order of [Al]-MFI > [Ga, Al]-MFI > [Ga]-MFI, indicating that the substitution of Ga for Al weakened the acidity. Then, we concluded that Ga is successfully incorporated into the zeolite framework. The amount of Brønsted acid of [Al]-MFI decreased after Zn2+ ion exchange. Meanwhile, the amount of Brønsted acid of [Ga]-MFI did not change after Zn2+ ion exchange, indicating that proton near Ga sites is hardly exchanged. This result suggested that proton near Al sites of [Ga, Al]-MFI was selectively exchanged with Zn2+ ion. The conversion of ethane and the BTX selectivity on EDA reaction were shown in Figure 1a and 1b, respectively. The catalyst lifetime has been successfully improved because the deactivation by coke deposition on the strong acid sites was suppressed by substitution of Ga for Al.
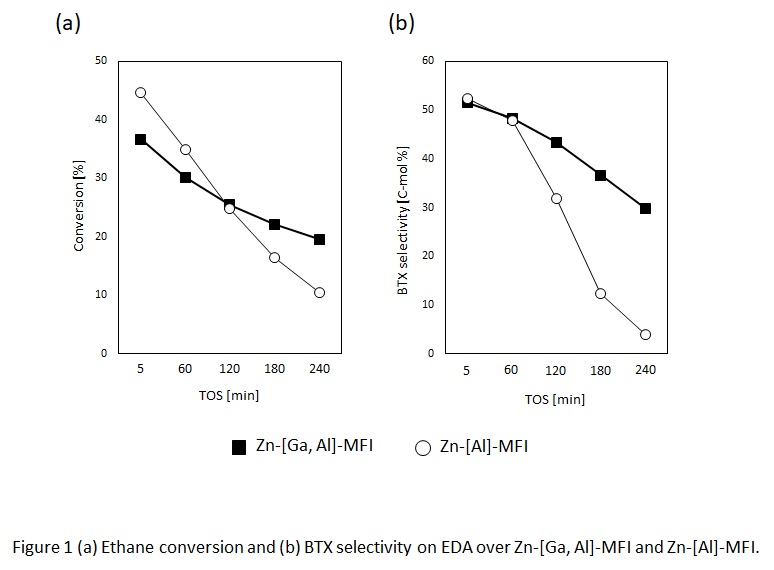
It was previously reported that the gas-phase one-step oxidation of benzene to phenol was carried out by using Cu/HZSM-5 modified with Ti as the catalyst. In this case, the almost same yield and selectivity of phenol was obtained by direct oxidation of benzene in comparison with the Cumene method. However, the morphology of Cu/HZSM-5 catalyst modified with Ti was the fine particle and the utilization of this morphology was not benefiting industrially. Cu/HZSM-5 was prepared by the Cu species impregnating on HZSM-5 which was one of zeolite. Although the Cu species was the active sites in this reaction, phenol di not produce when using supports other than HZSM-5. Therefore it was desire to prepare the forming of HZSM-5 zeolite, which had high thermal stability and did not use a binder. In this study, it is synthesized the binderless HZSM-5 (BLHZ5) following the preparation method of the patents from Nippon Shokubai and prepared the Cu catalyst doped on binderless HZSM-5 (Cu-BLHZ5) by an ion exchanged method.The structure and character of BLHZ5 supports which were prepared by hydrothermal synthesis method for different treatment times were investigation by XRD measurements and N2 adsorption-desorption measurements. From Fig. 1, it was cleared that XRD spectra of the BLHZ5 support indicated the formation of MFI structure for all hydrothermal synthesis time, even at 4h. On the other hand, the surface area and pore size of BLHZ5 were measured by N2 adsorption-desorption methods. No significant change of micro pore size for BLHZ surface area was observed for different synthesis time. However, the increasing of the BLHZ5 surface area was observed with increasing the synthesis time. Therefore, it is estimated that the optimum points of synthesis time is 72h.
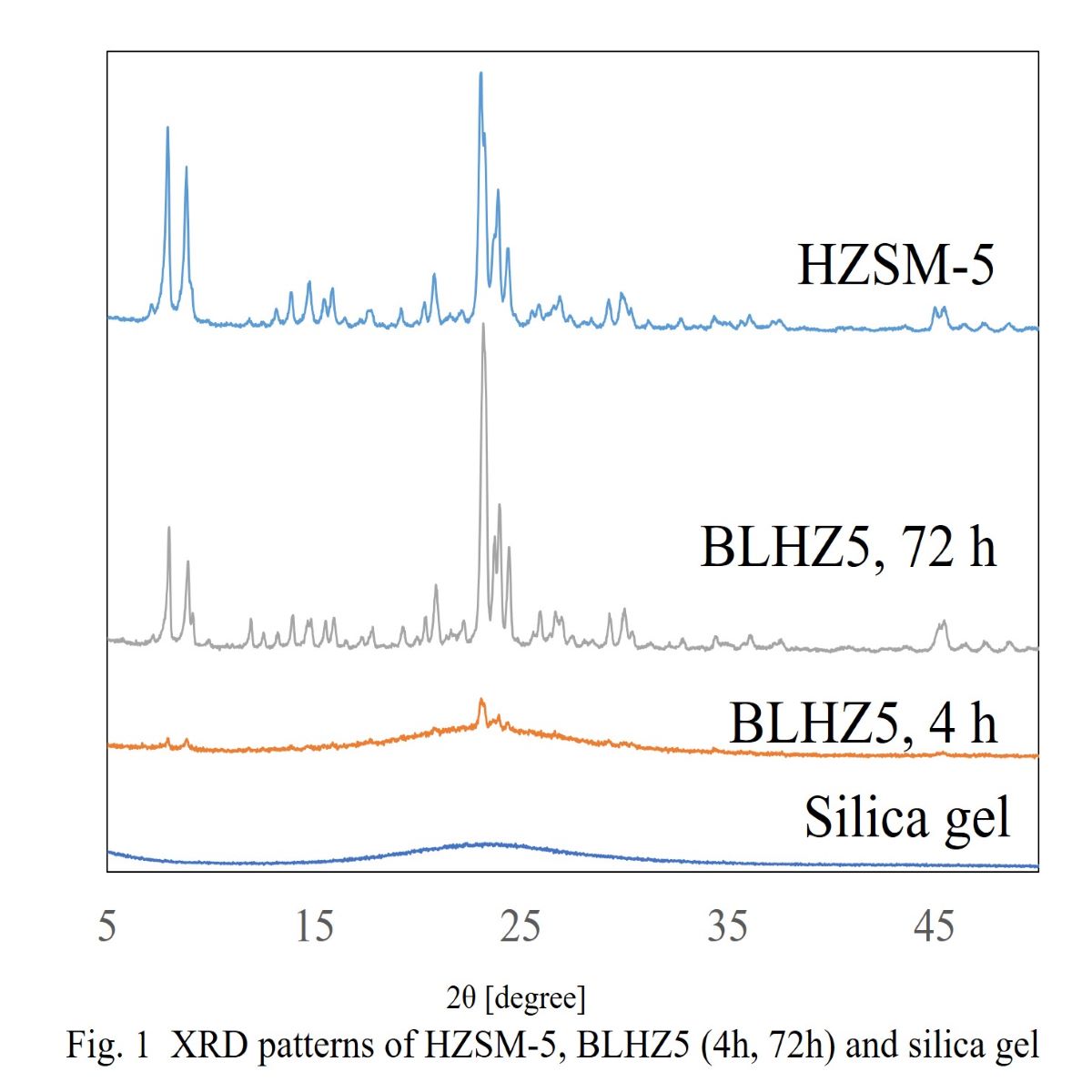
The reactive adsorption desulfurization (RADS) of a model gasoline n-hexane containing thiophene was conducted over a NiO/ZnO-Al2O3-SiO2 adsorbent in N2 and H2, respectively. A declining RADS trend has been observed in N2, without the presence of H2, indicating that NiO is sulfurized and exhibits activity for RADS. TPR and XPS results presented NiO in the adsorbent is difficult to be reduced because of the strong interaction between NiO and the support. The sulfurization of NiO into NiSx is a primary condition for RADS process the same as the presulfurization of hydrotreating catalyst, while metallic Ni is an intermediate reduction product of NiSx. The unreduced NiO/ZnO-Al2O3-SiO2 adsorbent performs a better desulfurization than the reduced adsorbent at the beginning of desulfurization process. NiO is assumed as the main active component and present a good desulfurization ability in RADS. Various pretreatments of the adsorbent showed the participation of hydrogen and n-hexane in pretreatment conducted at 420 °C contributed to the activation of adsorbent and the improvement of desulfurization performance under the reaction temperature of 300 °C. At last, some change of RADS mechanism is presented and discussed.
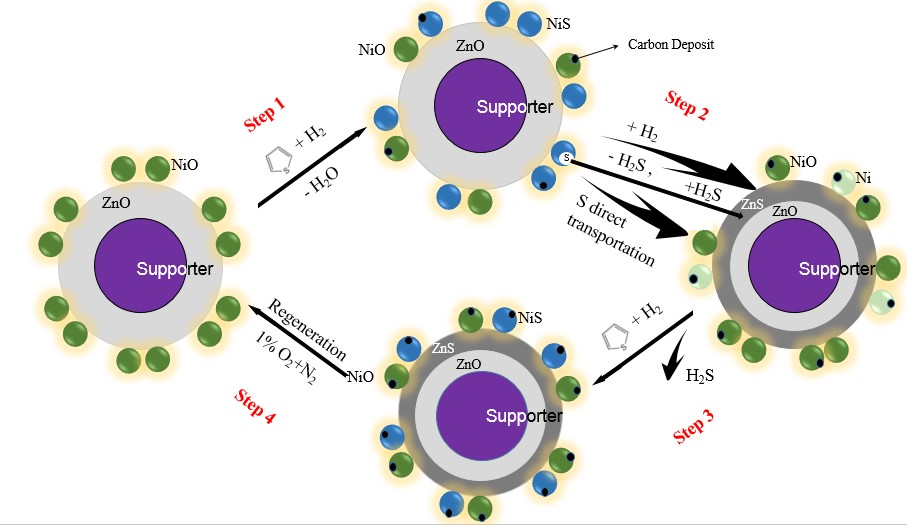
Methane steam reforming reaction is used to obtain hydrogen for use in fuel cells. In general, catalyst poisoning by the sulfur compounds is one of the main causes of catalyst performance degradation. Sulfur durability of alpha-alumina supported precious metal catalysts for methane steam reforming reaction were investigated. Methane conversion of all catalysts decreased in addition of tertiary-butylmercaptan (TBM) as a sulfur impurity in the reaction gas. Methane conversion of Ru catalyst was eventually deactivated almost completely in methane steam reforming with TBM. The time until catalyst deactivation depended on the amount of precious metal loading, and was further proportional to the sulfur adhesion amount and CO unit adsorption amount. In the TBM addition test of Rh catalyst, the methane conversion decreased to some extent, but thereafter the methane conversion reached a plateau and became a constant value. The methane conversion at the plateau was dependent on the loading of precious metal and the reaction temperature. There was a difference in adsorption characteristics with sulfur depending on the precious metal species. The performance degradation by sulfur poisoning in methane steam reforming reaction behaved differently depending on the precious metal species and their physical properties.
Nitrous oxide (N2O) is one of the greenhouse gases that lead to depletion of the ozone layer. Therefore, effective decomposition of N2O is imperative. In this study, MOR zeolite-supported Fe (Fe-MOR) catalysts were prepared by the various methods, and the effect of the preparation method on the N2O decomposition activity of the catalysts was investigated.
The Fe-MOR catalysts were prepared by the liquid and solid-state ion-exchange methods, using iron (III) nitrate enneahydrate (Fe(NO3)3·9H2O) and MOR zeolite as the support. In the liquid-state ion-exchange (LS) method, the MOR zeolite was immersed in Fe(NO3)3·9H2O aqueous solution and stirred at 80 °C for 3 h. After stirring, the ion-exchanged zeolite was filtered and washed, followed by drying at 110 °C for 12 h. This process was repeated arbitrarily. The dried sample was calcined at 500 °C for 3 h. In the solid-state ion-exchange (SS) method, the MOR zeolite was ground with Fe(NO3)3·9H2O in a mortar for 30 min. The mixture was dried at 110 °C above 12 h, after which, the dried sample was calcined under the same conditions as in the LS method. The Fe-MOR catalyst that was prepared by the LS process, which was repeated 4 times using Fe(NO3)3·9H2O aqueous solution (Fe/Al = 0.25), was denoted as LT-0.25-4. N2O decomposition was carried out using a fixed bed flow reactor at 500 °C.
The order of the N2O decomposition activity of catalysts used the same amount of Fe is as follows: SS-1-1 > LS-0.25-4 > LS-0.5-2 > LS-1-1. It is assumed that the Fe-MOR catalyst that was prepared by the SS method was not washed after grinding. Thus, much of the Fe species remained on the cation exchange sites, and this led to higher activity.
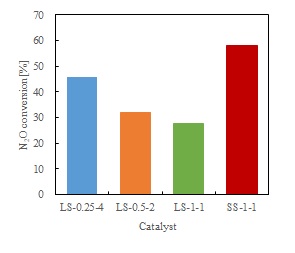
Incorporating covalently bound organic moiety to the surface of SiO2 is a general approach to tailor its surface properties for adsorbents and catalysts. Examples include tuning surface hydrophobicity/hydrophilicity by incorporating alkyl groups with various lengths, tailoring surface functionality by incorporating amine- and sulfonic acid-containing groups, and controlling interlayer spacing of layered silicates by grafting with alcohols. Functionalization of amorphous silica provides a cost-effective advantage. However, it involves a general challenge to achieve site-specificity and control a microscopic environment around functional groups. In contrast, connectivity defects (e.g. SiO–) contained in pure-silica zeolitic materials offer opportunities to achieve precise control of material properties. The location of connectivity defects in crystals is dictated largely by the local structure of zeolitic materials as well as the crystallographic location of charge-compensating structure-directing agents used in the synthesis of zeolitic materials. We chose lamellar precursors of a pure-silica MWW-type zeolitic material because these materials are known to possess abundant connectivity defects near the surface of zeolitic nanosheets and allow incorporation of bulky species between zeolitic nanohseets. Here, we report the site-specific functionalization of these materials with alcohols and alkanolamines and application of functionalized materials to the synthesis of metal-encapsulated catalysts.
Saccharification is a key step for the conversion of a main component of lignocellulosic biomass, cellulose, to chemicals and fuels. Enzymatic hydrolysis is the most popular saccharification method but suffers from disadvantages from an economic perspective since it is expensive and time-consuming. A potential alternative method is herein proposed, where cellulose is first pyrolyzed to anhydrosugars such as levoglucosan, and then anhydrosugars are hydrolyzed to glucose. Pyrolysis is a very fast reaction and does not require any catalyst or solvent. The yield of anhydrosugars reaches more than 90%, though it depends on reaction conditions significantly. It has been known that anhydrosugars are readily hydrolyzed to glucose in an acidic aqueous solution. Sulfuric acid is often employed as the catalyst. However, such homogeneous catalyst generates undesirable waste stream and is difficult to be recycled to the process. The present study demonstrates the availability of a solid acid catalyst for this reaction. A type of strongly acidic cation exchange resin was employed as the catalyst, and a main study was performed with levoglucosan as anhydrosugar model compound. The catalysis was very selective, producing glucose with near-complete yield. When compared to sulfuric acid at the concentration equivalent to the proton concentration in the test with solid acid, the reaction rate was comparable, and, moreover, the selectivity was better. The reaction rate was not altered by the concentration of levoglucosan in the range between 0.1 M and 0.5 M. The catalyst was also active toward cleavage of glycosidic linkages, which was confirmed in the test with cellobiose. Finally, the catalytic test was carried out with biooil from cellulose pyrolysis and successfully produced glucose with sufficient yields, based on anhydrosugar concentration of the oil, even in the presence of various other organic compounds.
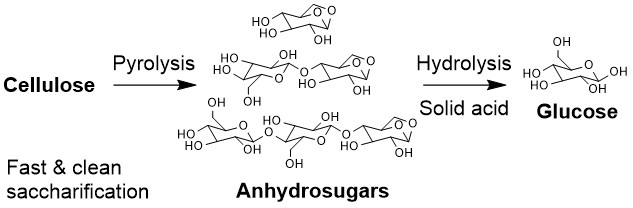
Caffeic acid phenethyl ester (CAPE) is a pharmaceutical ester with anti-cancer activity. It is recovered from propolis but is very expensive because of its low content (about 0.1%). On the other hand, esterification of caffeic acid and transesterification of chlorogenic acid (ester of caffeic acid and quinic acid) have been reported as chemical synthesis methods. Both caffeic acid and chlorogenic acid are contained in coffee beans at a relatively high concentration (approximately 8%) and these remain in spent coffee grounds (SCG). There is a possibility that CAPE might be synthesized from SCG. We have reported that ion-exchange resins show high catalytic activity of ester synthesis in non-aqueous system. In this study, we investigated whether CAPE was produced from SCG by ion-exchange resin catalysts.
First, extraction of caffeic acid and chlorogenic acid from SCG was performed using a conventional Soxhlet extractor. No caffeic acid was detected in the extract, but the chlorogenic acid content was about 0.6%, which was higher than that of propolis. Next, batch experiments of the CAPE production were performed using two kinds of ion-exchange resin catalysts. The resin catalyst was added to the reaction solution in which the extract was dissolved in phenethyl alcohol, and well shaken at 70°C. In case of using porous strong base anion-exchange resin, CAPE was not formed, which meant that the transesterification of chlorogenic acid did not proceed. In case of using porous strong acid cation-exchange resin, however, the CAPE concentration gradually increased as shown in Fig.1. Two sets of results under the same conditions overlapped each other, indicating a good reproducibility of the experiment. In addition, the CAPE concentration became higher than the initial value of chlorogenic acid. CAPE was considered to be formed from caffeic acid residues in other substances as well as in chlorogenic acid.
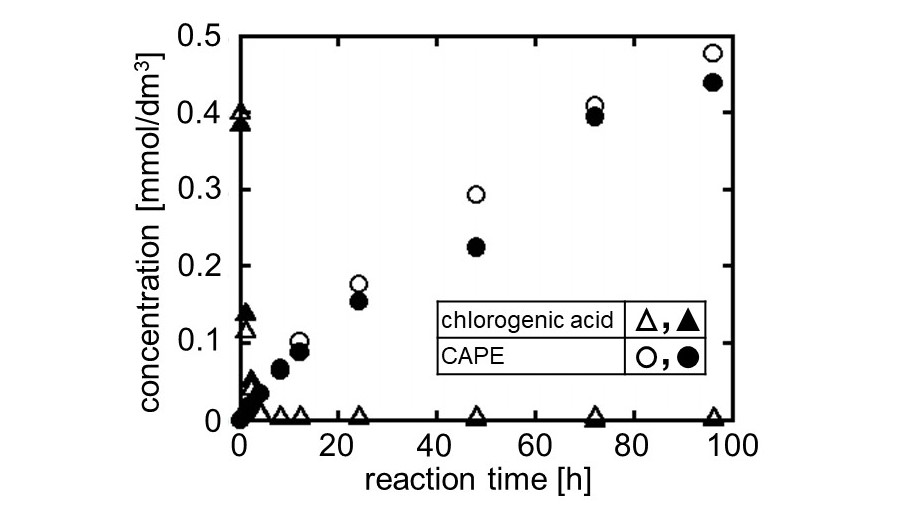
Undaria pinnatifida, brown macroalgae native to northwest Pacific areas, has been considered as a successful invasive species and is feared to cause a reduction in diversity and richness of native macroalgae. Though management of these invasive species is being investigated, it is often faced with failures which call for a resourceful utilization way of U. pinnatifida in a cheap and less energy-intensive way. In this study, U. pinnatifida is focused on as a feasible source for the production of solid acid catalyst. Solid acid catalyst from U. pinnatifida was prepared using sequential carbonization methods of pyrolysis and hydrothermal carbonization (HTC), and the influence of the combined carbonization methods on the characteristics of solid acid catalysts was investigated. The evaluation of catalytic performances of prepared catalysts was carried out using the esterification reaction of acetic acid and ethanol. Though catalysts prepared via HTC method without pyrolysis pretreatment showed better acidity, it had less porosity. Catalysts prepared with pyrolysis pretreatment showed higher specific surface area and coherently showed higher catalytic activity. When the catalytic performance was compared with conventional catalyst Amberlyst-15, U. pinnatifida derived solid acid catalyst showed higher conversion rate of acetic acid and ethyl acetate yield. Through this study, it is shown that marine biomass, U. pinnatifida can function as a solid acid catalyst with good catalytic performance and favorable reusability profile.
Lignin, a polymer with alkyl phenol units, has attracted attention as a resource for phenols. Our previous study has suggested a lignin conversion process to produce monomeric phenols from lignin. The degraded lignin that was depolymerized in water/1-butanol mixed solvent, was converted to monomeric phenols over TiO2-FeOx catalyst with high activity for oxidative decomposition and demethoxylation of lignin units. However, the catalyst was not effective for cleavage of C-C bond between the units of lignin structure. Thus, this study investigated the catalytic reaction of the lignin over a mixed catalyst of TiO2-FeOx and H-ZSM-5 for C-C bond cleavage.
3 wt% of degraded lignin in quinoline solvent was used as feedstock because quinoline was found to be inactive over the catalyst, and also had a good solubility against the degraded lignin, which was consistent with the investigation based on Hansen solubility parameters. The reaction was carried out using a fixed-bed flow reactor at 673 K for 2 h under atmospheric pressure.
As a result, TiO2-FeOx catalyst exhibited only 1.7 mol% of phenols from the lignin. H-ZSM-5 exhibited only 0.5 mol% of phenols, meaning that H-ZSM-5 was not effective for oxidative decomposition. Moreover, the degraded lignin molecular size was too large compared with pore size of H-ZSM-5 that the molecules could not diffuse into the pore and be decomposed. On the other hand, the physically mixed catalyst was high activity for recovering monomeric phenols from the lignin, achieving 11.7 mol% of phenols. Furthermore, GPC chromatogram indicated that molecular weight of products in the mixed catalyst was lower than that of products in only TiO2-FeOx or H-ZSM-5 catalyst individually. The mixed catalyst could dissociate C-C bond in the degraded lignin, leading to the improvement of the yield of phenols. In addition, the between the type of zeolite and reaction activity will be reported.
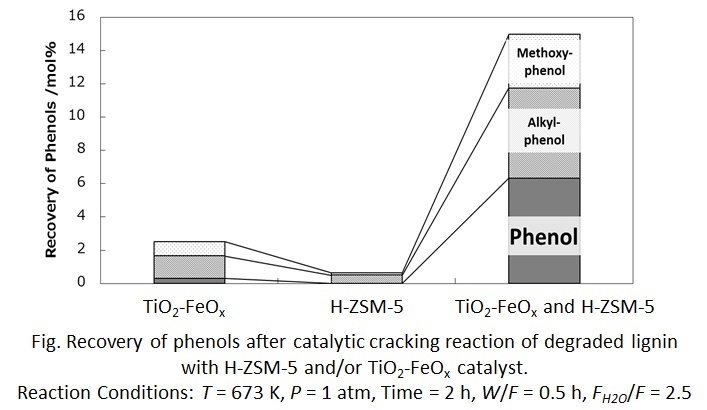
Lignin valorization is one of the biggest challenges for developing biomass-based chemical industry. Recent innovative works has enabled depolymerization of lignin to phenolic monomers with the yield close to the theoretical maximum. Hydrogenation in alcohol with heterogeneous catalysts is an example of the effective methods. The product from such effective depolymerization is a mixture of phenols derived mainly from the cleavage of β-O-4 linkages between phenylpropanoid units. However, the obtained highly functionalized phenols are not common in current industry and, therefore, needs further processing for utilization in the intended applications. The present study shows that simple phenols without functional groups at para-position form selectively in lignin depolymerization in hot compressed water with bases, such as sodium carbonate, as a type of catalyst. Alkali lignin derived from hard wood and bamboo released guaiacol and mixture of phenol, guaiacol, and syringol, respectively, as the main phenolic products. The selectivity to these phenols were more than 60% among identified phenols. The total phenols yield was not so high, up to 9%, but the selective formation of simple phenols was unusual when considering that the weakest and most abundant linkage was β-O-4. Although the reaction mechanism has not yet been clear, analysis of remaining lignin at different reaction times with several techniques showed that the lignin was condensed rather than depolymerized while releasing the phenols. Temporal change in the phenols composition indicated that the simple phenols were directly released from the lignin. On the other hand, utilizing this findings, the study also demonstrated a process that produced the simple phenols and high calorific value fuel gas along with near-complete recycle of the base catalyst.
Pyrolysis of cellulose has been studied for investigating combined effects of operating variables such as heating rate, type of reactor and a structural property of cellulose (crystallinity) on characteristics of its conversion into volatiles and char. A commercially available microcrystalline cellulose (MCC; crystallinity index (C.I.) = 87%) was subjected to ball-milling, which amorphized the MCC to C.I. of 60% and finally 0%. The MCC and amorphized celluloses were pyrolyzed at 445–764 °C with heating rates of around 103 °C/s in a Curie-point reactor where the secondary homogeneous and heterogeneous secondary reactions of volatiles were minimally involved. Results of the pyrolysis revealed two important characteristics. Firstly, the char yield was even below 1 wt% when the peak temperature was 590 °C or higher, and was much less than that from slow pyrolysis in a thermogravimetric analyzer, ca. 10 wt%. Suppression of the secondary reaction of the volatiles was thus effective for avoiding the char formation. Second, the char yield was almost independent of the crystallinity. The yield of the key product, H2O, formed by cross-linking, was hardly influenced by the crystallinity. The above-mentioned findings lead to a novel concept of char-free fast pyrolysis of any types of cellulose to produce mono- di- and oligo-anhydrosugars at high yield within seconds.
Biomass gasification process needs to be developed to ensure the future energy system. The important issue of the gasification system is to remove tar component generated in the gasification process. In order to achieve this purpose, a suitable catalyst should be developed for eliminating tar by steam reforming. Our group have already developed a powerful structured Ni/Al2O3 catalyst for steam reforming of toluene or naphthalene as the main components of tar [1][2]. In this study, we investigated the properties of the structured Ni/Al2O3 catalyst for simultaneous steam reforming of toluene and naphthalene mixture as to evaluate a viability for the practical application of the structured catalyst to tar reforming [3].
The structured Ni/Al2O3 catalyst was prepared by a sol-gel method and an electroless plating [3]. After H2 reduction at 800 °C for 1 h, the performance test was evaluated under the following conditions: setting temperature was 800 °C-830 °C; feed rate of toluene or mixtures (toluene/naphthalene=9/1) were 9.6×10–4 mol·min-1, and feed rate of naphthalene was 9.6×10–5 mol·min-1; steam to carbon ratio (S/C) was 1.5.
Figure shows reforming conversions for steam reforming of toluene (SR-T), naphthalene (SR-N) and mixtures of toluene and naphthalene (SR-TN) over the structured Ni/Al2O3 catalyst. In case of SR-T and SR-N, both reforming systems showed a high activity at 800 °C: each conversion was almost 100%. On the other hand, mixed reforming system showed deactivation in short time at 800 °C: the conversion was decreased from 92.9% to 33.6% during 3 h. Considering that reactivity of toluene is higher than naphthalene [4], naphthalene might not be sufficiently reformed and converted into a carbon precursor. Such serious problem was solved by setting reforming temperature at 820 °C or 830 °C. This result suggested a progressive advantage of Ni-based structured catalyst with high heat transfer ability.
Gasification of woody biomass can provide synthesis gases (CO, H2) at low temperature, however, byproducts such as tar, char, H2S, and NH3 are also generated at the same time. As these byproducts affect downstream equipment operation and reduce total energy efficiency, they should be treated appropriately before entering the downstream equipment. From this background, our research group have been proposed a new consecutive gasification and purification process that is consisted of biomass gasification, cyclone for char collection, metallic filter for volatile minerals, selective H2S decomposition, simultaneous decomposition of tar and NH3, and selective CO2 absorption. While the consecutive gasification and purification processes have been studied in our research group for a long time, developing CO2 selective absorbent material for produced gases is still remained as a research topic. Therefore, this study was started as a first step for developing selective absorbent material. At first CO2 absorption basic condition (material loading amount, pretreatment method, reactant flow rate, temperature, inner size of reactor, etc.) for obtaining the CO2 breakthrough profiles with all absorbent materials was determined. Then CO2 breakthrough profile was obtained with each sample under 10%CO2/Ar mixture flow, and the most excellent absorbent material was selected in terms of CO2 absorption capacity. In addition, both temperature and time needed for regenerating the used material was investigated using XRD and TG-DTA measurements. Finally, selected material was applied for CO2 absorption-regeneration cycle test to see the possibility of recycle usage.
With the aim of efficient use of triglycerides as alternative fuels, deoxygenation of them and conversion to hydrocarbons are important for improving combustion heat. The basic approach of this conversion is a hydrodeoxygenation process, but using a pressurized hydrogen atmosphere incurs high process cost. Alternatively, we focused on a fluid catalytic cracking (FCC) process that can convert triglycerides to hydrocarbons producing CO, CO2, and H2O without using hydrogen atmosphere. Here, the deoxygenation pathway producing H2O is desirable for efficient conversion because the deoxygenation producing CO or CO2 cause a carbon loss. However, the detailed mechanism of H2O formation pathway has not been understood well. This study focused on the effect of a hydrogen transfer reaction that proceeds in the FCC process and investigated the reaction mechanism of ester deoxygenation using deuterated reagents as a hydrogen donor.
Ethyl hexanoate (ester model compound) and deuterated n-hexadecane (n-C16D34, hydrogen donor model compound) were mixed with an FCC equilibrium catalyst. The mixture of feedstocks and catalyst was instantaneously heated to 500°C using a Curie point pyrolyzer, and the reaction products were analyzed using a GC-MS. Figure 1 shows the mass chromatogram of the produced water in the catalytic cracking of ethyl hexanoate/n-C16D34 mixture compared with that of ethyl hexanoate/n-C16H34 mixture. In the reaction with n-C16D34, formation of HDO as well as H2O was observed whereas that of D2O was hardly observed. This result suggests that the ester received one hydrogen atom from the hydrogen donor and produced H2O. Thus, it was indicated that the hydrogen transfer reaction contributes to the deoxygenation of triglyceride, which can be useful information for a catalyst design with efficient conversion of triglycerides to hydrocarbon fuels.
Woody biomass has high oxygen content and requires an efficient deoxygenation process for upgrading to hydrocarbon fuel. Co-processing of bio-oil in the fluid catalytic cracking (FCC) of heavy oil is one of the effective methods for that purpose. Our recent study revealed that the hydrogen transfer reaction in the FCC can accelerate deoxygenation producing water rather than CO2 and CO evenunder a hydrogen-free atmosphere, which is an efficient deoxygenation pathway avoiding carbon loss. However, the effect of bio-oil property, which depends on the type and condition of the liquefaction pretreatment, on the deoxygenation pathway and the product composition in the co-processing FCC has not been understood well.
The previous study reported that the co-processing FCC of bio-oil prepared with fast pyrolysis resulted in a large amount of coke formation and low liquid yield. This may be due to the formation of condensed products during the pyrolysis that were difficult to upgrade in the FCC process. In this study, we focused on the solvolysis liquefaction that can produce bio-oil with high liquid yield (~90%) at a milder temperature than pyrolysis, thus avoiding the condensation reaction.
Bio-oil was prepared from the solvolysis of palm kernel shell (PKS). Catalytic cracking of the mixture of bio-oil and tetralin (heavy oil model compound) was investigated using a Curie point pyrolyzer connected directly to a GC-MS, and compared with the reaction of the mixture of powdered PKS and tetralin. The catalytic cracking of the bio-oil/tetralin mixture showed higher H2O/(CO2+CO) and Monoaromatics/Polyaromatics (3+ring aromatics) ratios than that of the powdered PKS/tetralin mixture. This result suggests that the pyrolytic reaction should be avoided through the whole process of liquefaction and catalytic cracking for the efficient conversion of the woody biomass, and the solvolysis pretreatment is adequate for that prosess.
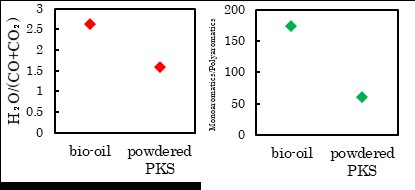
Microalgal biomass has high productivity and expected as an alternative fuel resource. Euglena gracilis is one of such promising microalgae, whose mass cultivation in an open pond has been achieved recently. During the many steps of the fuel production from microalgae, oils are extracted from algal cells, purified and converted to transportation fuels. The most common conversion method is transesterification to biodiesel, but the biodiesel is an oxygen-containing fuels whose heat value and thermal stability are lower than that of hydrocarbon fuels. Conversion to hydrocarbons are usually conducted with hydroprocessing, but using pressurized hydrogen atmosphere requires high operation cost. Then, we focused on the fluid catalytic cracking (FCC) process, which is the major oil refinery process that can upgrade heavy oils to light fractions without using hydrogen atmosphere. In our previous study, we confirmed that the FCC process can convert wax esters derived from Euglena gracilis to hydrocarbons fuels with efficient deoxygenation. Furthermore, the FCC process is expected to be resistant against the impurities because it uses a circulating fluidized bed reactor and the catalyst is regenerated continuously in the regeneration tower. Therefore, in this study, we investigated the catalytic cracking of unrefined microalgae oil and compared the reaction with that of refined oil.
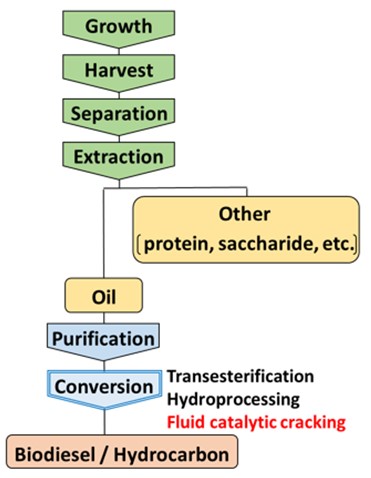
Polyisobutylene (PIB) manufactured by the living cationic polymerization exhibits desirable properties such as thermal stability and high barrier properties. Living polymerization often requires quite low reaction temperatures to avoid termination and chain transfer, for example, the polymerization of PIB is operated at around -80 to -70 °C in batch system. Living cationic polymerization of isobutylene (IB) utilizing TiCl4 in the batch system at low temperatures has been investigated [1-3]. It has been proposed that cationic reactive chain ends are generated in the equilibrium between dormant (tert-alkyl chloride chain ends) and active (ion-paired chain ends). However, the whole reaction mechanism remains to be elucidated due to the difficulty of direct evaluation of concentrations of the dormant and active species in the equilibrium. Moreover, understanding of the reaction mechanism is a key for process intensification.
Quantum chemical approaches have been used well to investigate polymerization reactions [4, 5]. These approaches have been showed the details of chemical reactions such as molecular geometries of reactants, products and transition states, as well as the changes of enthalpy, entropy and Gibbs free energy. The information provided by quantum chemical studies have helped not only to better understand reaction mechanisms, but also to design more efficient reaction processes.
In this work, we report the investigation of the reaction mechanism of the ionization equilibrium in the PIB polymerization with several catalysts using quantum chemical calculations. Experimental results of PIB polymerization with different catalysts are also discussed.
[1] Storey, R. F., Choate, K. R., Jr., Macromolecules, 30, 4799-4806 (1997)
[2] Fodor, Z., Bae, Y. C., Faust, R., Macromolecules, 31, 4439-4446 (1998).
[3] Tawada, M., Faust, R., Macromolecules, 38, 4989-4995. (2005)
[4] Dossi M., Storti G., Moscatelli D., Polym. Eng. Sci., 51, 2109 (2011)
[5] Deglmann P., Muller I., Becker F., SchaferA. , Hungenberg K.D., Weiß H., Macromol. React. Eng., 3, 496 (2009)
A kinetic model was developed for the synthesis of methyl acetate (MA) from dimethyl ether (DME) over ferrierite catalyst. Two types of active sites were considered; zeolite-exchange site that stabilizes acidic protons and methyl and acetyl groups, and CO-binding centers for non-competitive binding of DME and CO (Cheung et al., J. Catal., 245 (2007) 110–123). Pseudo-steady-state approximation was applied to develop the reaction rates, and kinetic parameters were estimated by fitting experimental data under a variety of temperature, pressure, space velocity, and feed composition. The developed model was used to evaluate the effects of operating conditions on the conversion and selectivity.
Clean and efficient utilization of low-rank coal is of great significance to the sustainable development of energy in China. “Regenerative Calcium Carbide Production Process” was proposed for clean and efficient utilization of low-rank coal. This technology takes pulverized medium to low rank raw coal and raw lime as raw materials that can form pellets. Then the pellets were sent into sealed calcium carbide furnace after pyrolysis in preheating furnace to obtain calcium carbide and pyrolysis oil and gas at the same time. The pyrolysis process of pellets is the key step of the technology. A comprehensive analysis of the influence of lime addition, pellet properties and operation conditions on pellet pyrolysis process plays an important role in comprehensively understanding pellet pyrolysis process and optimizing this new technology.
Firstly, the thermogravimetric experiments and basic experiments of pellet pyrolysis in fixed bed were carried out. The effects of calcium oxide, heating rate, pyrolysis temperature and pyrolysis time on the pyrolysis process of pulverized coal were investigated. The experimental data were provided for the establishment of pyrolysis kinetics. The results show that the addition of calcium oxide, the increase of heating rate, pyrolysis temperature and pyrolysis time can increase the volatile in the pyrolysis of pulverized coal. Then, the double distributed activation energy model based on genetic algorithm is chose to establish the pyrolysis kinetics. The results show that the double distributed activation energy model based on genetic algorithm can accurately describe the dynamic process of pellet pyrolysis. By comparing the pyrolysis kinetics, it is found that the addition of calcium oxide can reduce the activation energy of pulverized coal pyrolysis and increase the reaction rate of coal pyrolysis.
Meerwein-Ponndorf-Verley (MPV) reduction can selectively produce unsaturated alcohols from corresponding unsaturated aldehydes by using secondary alcohols as a hydrogen donor. Recently, bio-alcohols are expected to be used for MPV reduction. In bio-alcohols, large amount of water is included, and leads to decrease in the catalytic activity of Zr supported catalysts for MPV reduction due to the strong adsorption of water over Lewis acid sites on Zr species. In this study, MPV reduction of cinnamaldehyde to cinnamyl alcohol was carried out over Zr supported catalysts using activated carbon (AC) as supports possessing the hydrophobic properties.
Zr/AC catalysts were prepared by an impregnation methods. Zr loading was 10 wt%. In this study, 20 kinds of ACs were employed as supports of Zr/AC catalysts. Surface area of micropores and mesopores was determined by means of N2 adsorption isotherms. MPV reduction of cinnamaldehyde was carried out in an autoclave under 1 MPa (gauge) of N2 at 453 K for 1 h in 2-propanol with or without water (12.5 wt%).
To evaluate the hydrophobicity of each Zr/AC catalyst, water resistance was defined as cinnamyl alcohol yield in the presence of water divided by that without water. Over the Zr/AC catalysts possessing the mesopore surface area greater than 100 m2/g, the lower levels of water resistance were observed, suggesting that Zr species over the surface of mesopore was strongly poisoned by water. With respect to the Zr/AC catalyst indicating the highest water resistance, Zr species was selectively loaded in the micropore of activated carbon. These results suggest that the porous structures in AC strongly affect the water resistance of Zr supported AC catalysts.
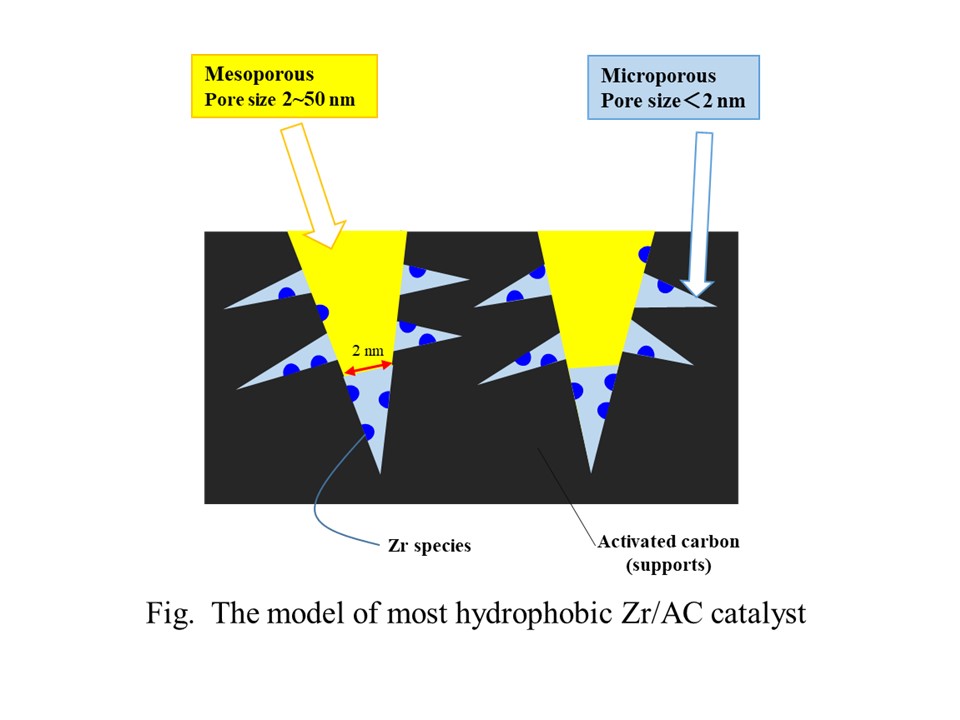
Short-chain fatty acids (SCFAs) have been used as raw materials in wide range of chemical and medical applications. One technique to produce SCFAs is oxidative cleavage of long-chain fatty acids (LCFAs). However, unless the LCFAs are unsaturated, the yield of SCFAs is often very low because the carboxylic group of the fatty acid is more active than other part of the molecule. This work explores the idea of introducing a double bond into saturated LCFA, i.e., stearic acid, via selective dehydrogenation using commercial heterogeneous catalysts. However, cracking of the LCFA is also catalyzed. Different type of metals was therefore investigated to study the effect of metals on the cracking and dehydrogenation. The experiments were conducted in an autoclave reactor under inert atmosphere. The temperature was in the range of 250-350 degree celsius. The products were analyzed by gas chromatography equipped with mass spectroscopy (GC/MS). The results reveal that the introduction of double bond in the aliphatic chain of the stearic acid is possible although the yields of the unsaturated LCFAs are low. Effects of various parameters, such as temperature, pressure, and reaction time, were also investigated and reported.
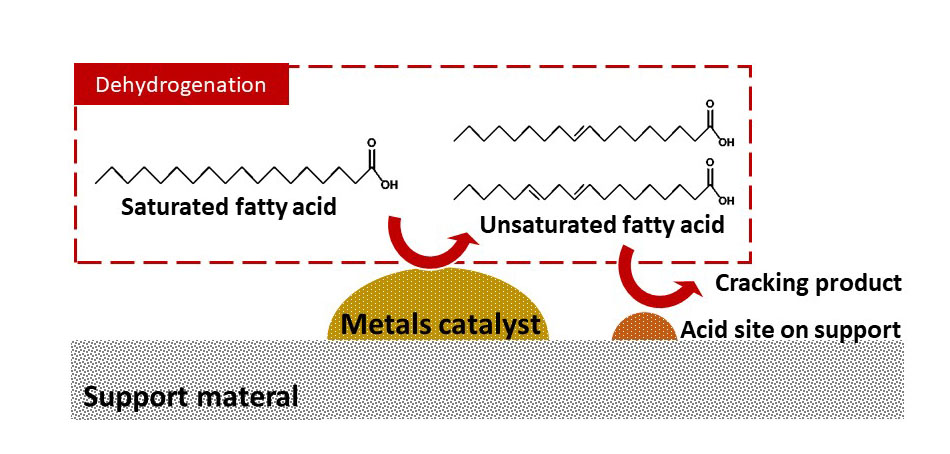
Short-chain fatty acids (SCFAs) and medium-chain fatty acid (MCFAs) are valuable raw materials in wide range of chemical and medical applications. They can be converted to other derivatives by known chemical reactions. Unfortunately, both SCFAs MCFAs are not as abundant in nature as long-chain fatty acid (LCFAs). In this work, the oxidative cleavage of oleic acid, which is one of the most abundant unsaturated LCFAs in nature, was studied. The oxidation was induced by hydrogen peroxide catalyzed by a commercial alumina-supported metal catalyst. The products were analyzed by gas chromatography equipped with mass spectroscopy (GC/MS). The results revealed that MCFAs and SCFAs were detected in a product. In addition, aldehydes were also found. In this work, effects of catalyst loading, oleic acid-to-hydrogen peroxide ratio, reaction time were investigated and reported.
Lactic acid is produced by the decomposition of sugars by lactic acid bacteria by fermentation, and is used as a raw material for biodegradable plastics.
Since the result of lactic acid fermentation depends on culture conditions such as sugar type and concentration, temperature and pH, it is necessary to set appropriate conditions depending on the bacteria used.
In this study, lactic acid fermentation using shochu lees without sterile condition at high temperatures was conducted to investigate effects of cultivation pH (5-7) and temperature (55,60 °C). Sludge collected from a pond on campus was used as inoculum.
The result was that most sugar was consumed and lactic acid was produced at the operating conditions of 55 °C and pH 6.5.
Many important chemical reactions, such as the water-gas shift reaction and ammonia synthesis via Haber-Bosch process, are reversible in nature and the conversion of product(s) limited by chemical equilibrium. Here, we have shown that by de-coupling the overall reaction into two or more sub-reactions via the use of a heterogeneous mediator (a reaction scheme known as “chemical looping”), it is possible to overcome the chemical equilibrium of the overall reaction and achieve almost full conversion of the products.
We demonstrate this possibility by performing the water-gas shift (WGS) reaction at 820 °C: If WGS is performed at this temperature using conventional reaction scheme with an equimolar feed of carbon monoxide and steam is used, the product stream will contain approximately equal amount of carbon monoxide, steam, hydrogen and carbon dioxide on a molar basis when equilibrium is achieved. On the other hand, by feeding the reactants counter-currently over a packed bed of solid mediator that can supply and uptake oxygen over an appropriate range of oxygen fugacities (rather than at a fixed oxygen fugacity), such as non-stoichiometric perovskites (and in this particular case, La0.6Sr0.4FeO3-δ), we are able to produce separate streams of high purity hydrogen and carbon dioxide separately, achieving average conversions over 80% for both carbon monoxide and steam.
The conversion that can be achieved depends on the oxygen fugacity – oxygen capacity relationship of the solid mediator and can be further increased by optimising the length of the bed and the cycle time (duration of each feed stream in the cyclic process). In theory it is possible to achieve full conversion of carbon monoxide to carbon dioxide in one half-cycle and steam to hydrogen in the other. In this way, not only have the equilibrium limitations been lifted, downstream separation units can be simplified or even eliminated.
A methanation processes using synthesis gas derived from naphtha, biomass or coal can generally be performed in both fixed and fluidized bed reactors. The fluidized bed methanation allows isothermal operation due to the high heat transfer coefficients, simple and easy control of the operation. Further advantage is that it is possible to easily remove, add and recycle catalyst continuously during operation. On the contrary, special attention should be paid to attrition and the entrainment of the catalyst particles. In the fixed bed methanation, the heat of reaction has to be considered because of the high amount of CO in the syngas. For controlling the temperature inside the reactor, several methanation reactors are connected in series with intermediate gas cooling or recycle of product gas.
However, the standards for natural gas pipeline network in Korea and Japan are calibrated in the range of 9,700 ~ 10,800 kcal/m3. In order to connect the SNG produced from coal to the pipe network, the calorific value should be adjusted by adding gaseous fuel such as LPG (Liquefied Petroleum Gas).
For this purpose, it is necessary to synthesize paraffinic hydrocarbons having C2 ~ C5 carbon number by designing a catalyst so that FT (Fischer-Tropsch synthesis) reaction occurs simultaneously with the methanation reaction. Therefore, in this study, the reaction characteristics to produce the higher calorific SNG (including C1 to C5 range hydrocarbon) over the hybrid catalyst were investigated in bench-scale fixed bed reactor (FBR), bubbling fluidized bed reactor (BFBR) and slurry bubble column reactor (SBCR).
Acknowledgement:
The authors are grateful to the financial support from Korea Institute of Energy Evaluation and Planning (KETEP, No. 20173010050110).
References:
Kang, S.H., Woo, K.J., Bae, J.W., Jun, K.W. and Kang, Y., Korean J. Chem. Eng., 26, 1533-1538(2009)
Among various methods to produce hydrogen from methane, catalytic direct decomposition of methane is the most environmental friendly process because COx is not generated. CH4 direct decomposition with the catalyst normally provides two hydrogen molecules and carbon deposited on the catalyst as an amorphous carbon or carbon nanotube or film-like carbon. After reaction, those carbonaceous species were separated from the catalyst and collected as functionalized carbon materials. In the most cases, a fixed bed reactor was used for this reaction, and the catalyst deactivation was observed after a short reaction time due to carbon deposition. Some research groups had reported that the usage of plasma or membrane reactor improved the catalytic performance for the direct decomposition of methane; however, catalyst deactivation was still observed, and the way for separating solid carbon from the catalyst and catalyst regeneration method are not clear. From these backgrounds, our research group recently proposed a newly circulating bed reactor which consists of cyclone type reactor for methane direct decomposition reaction and riser reactor for regenerating the carbon deposited catalyst with steam to produce synthesis gas. As a novel circulating bed reactor is under construction, methane direct decomposition with Fe-based or Co-based catalysts and steam gasification of the carbon deposited on the catalysts were performed in a fixed bed reactor in the present study. It was found from the obtained results that the catalytic activity for methane direct decomposition depended on metal loading amount and type of metal and support. Moreover, the kind of carbon deposited on the catalyst was changed with the aforementioned parameters. Since it could be predicted that the reactivity of those carbonaceous species with steam might be changed, the results obtained from steam gasification of carbon deposited on the catalysts were also presented.
Over last decades, electrochemical advanced oxidation processes based on Fenton reaction (EAOP-Fenton) have been paid great attentions because they are capable of continuous generation of strongly oxidizing radicals that can completely oxidize organic pollutants into CO2 and H2O. Among the EAOP-Fenton processes, Fered-Fenton (FF) process, which relies on the electrochemical reduction of Fe(Ⅲ), has demonstrated great effectiveness and has potential for practical application. In FF process, a variety of factors have an impact on the efficiency, although the cathode potential that is especially important because not only in-situ generation of Fenton reagent but competition reactions can occur on the cathode. Furthermore, many researchers have shown the utility of FF process, but most of them have referred to a batch reactor. It is required to develop a flow-type reactor, which achieve continuous treatment, for the application of FF as an industrial method. We have previously presented a flow-type reactor with macro porous carbon electrode as cathode, which is water-permeable. The electrodes were prepared through the carbonization of carbon particles molded with binder.
In this study, effect of reaction conditions such as flow rate, current, on the degradation ratio was examined to understand the electrochemical reaction in the electrodes. It was found that ionic transfer in the electrode is a key factor. Therefore, we measured the liquid potential in up and downstream of the cathode to elucidate the ionic transfer in the cathode. Anode electrode was placed in the downstream of the cathode. Liquid potential in the downstream side was raised with the increase of current, whereas that in upstream side was not changed, indicating that reaction does not occur on the electrode in the upstream side. Change of liquid potential in the cathode was also estimated using electrodes with different thickness.
As photocatalysis is one of useful methods for the cleanup of polluted water, various photocatalyst powders have been reported. Once photocatalyst powder was dispersed into the polluted water, clean water without any organic compounds could be obtained under light irradiation; however, the separation process is necessary to remove the powders from clean water, which imposes high cost and reduces the recovery rate. To solve these problems, our research group proposed a novel catalyst system that photocatalyst powder is encapsulated in alginate shell, and both reactants and products could be permeated through the shell by the concentration gradient between inside and outside. Usage of this system makes catalyst recovery easy, and catalyst-loaded capsules could be successively used several times if the capsules are not broken. In our previous study, two kinds of photocatalyst-loaded alginate capsules were successfully prepared and applied for the degradation of methyl orange under light irradiation. It was found that this new system operated smoothly, and encapsulated nitrogen doped TiO2 powder prepared by original sol-gel method showed higher activity than commercial TiO2-loaded capsules under the irradiation of ultraviolet and/or visible light. Characterization results obtained by various analytical methods suggested that catalyst surface area, crystallinity of anatase TiO2 phase, light absorption property, and powder dispersibility inside the capsule might be key factors to determine its activity. In this study, high surface area TiO2 powders were prepared by original sol-gel method and loaded into the alginate capsules. These capsules were also tested, and the performance was compared with the results obtained in a previous study. Finally, it is indicated which are significant factors to exhibit excellent performance for the degradation of methyl orange under light irradiation.
The reactant (carbobenzoxy phenylalanine) is soluble in an organic solvent and hydrogenated to the product (phenylalanine) using solid catalyst. Phenylalanine is not soluble in the organic solvent, so it needed to be dissolved in an aqueous phase. This was the gas-liquid-liquid-solid four-phase system. Mass transfer becomes more complicated in comparison to three-phase reactors as the catalyst needs to efficiently contact a gas phase as well as two immiscible liquid phases. The catalyst surface properties (hydrophilic versus hydrophobic) also play an important role in the reaction related to the wetting efficiency of the liquids at the solid interface. Gas-liquid-liquid cocurrent upflow reator was used. Column inner diameter was 2.0 cm. Gas flow rate was 1.7x10-6m3/s. Liqud flow rate ratio of water to organic solution was 3. 1-octanol was used as organic solvent. Reaction temperature was 323 K.
To improve Pd/Al2O3 (3 mm in diameter, 3.2 mm inlength) reaction rate, 0.3 mm glass beads were inserted to the catalyst bed. Object of glass bead was calming the gas and liquid flow patern and incresing the gas and liquid velocity.
Gas and liquids hold up was measured to determined the residence time. Water was the continuous phase in this reaction system. Each liquid residence time was calculated from holdup and flow rate. Total liquids flow rate were varied and conversion was measured in each liquid flow rate. Figure 1 shows the effects of the organic solvent residence time on the conversion. Two type of catalyst bed were used. Diluted bed is shown in green mark and catalyst only bed is shown in blue mark. Conversions were linealy increased with increase of the residence time. Slopes of the lines show the reaction rate. The raction rate was increased 1.6 times by using glass beads.
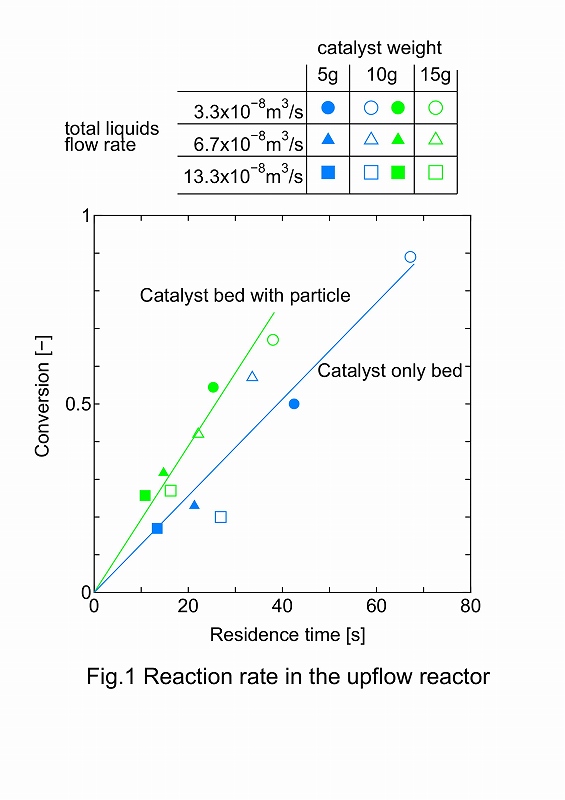
Zeolites are microporous crystalline aluminosilicates, and are industrially used as adsorbents and catalysts because of their very specific physicochemical properties such as ion exchange, adsorption properties.and solid acidity. CHA type (CHA) zeolite, which is one of the most important zeolite, has a three-dimensional pore system with ellipsoidal large cages (6.7x10 â) that are accessible via eight-membered ring windows (3.8x3.8 â). High silica CHA zeolite SSZ-13 and silicoaluminophosphate zeotype SAPO-34 with CHA topology are of great interest as they exhibit a particular shape selectivity for converting methanol or bioethanol to light olefins such as ethylene and propylene. Pure silica CHA also has potential for industrial applications such as adsorption and separation of organic molecules and gas storage because of a larger void space due to the absence of counter cations in the pores and the hydrophobicity. Recently, the hydrothermal conversion of one zeolite into another, i.e., interzeolite transformation has attractive interest from the standpoint of an alternative strategy for zeolite synthesis. Zones and van Nordstand have already reported the interzeolite conversions of cubic P and FAU zeolites used as only aluminum source to high-silica CHA zeolite SSZ-13 with Si/Al ratio of 13. In this study, we investigated influence of Si/Al ratio of FAU zeolite as silica and aluminum sources in the interzeolite conversion to high-silica CHA zeolite. As a result of detailed investigation, by using FAU type zeolite as a silica source and an aluminum source, the Si / Al ratio of the obtained SSZ-13 was successfully extended to 10 to 200. In addition, Moreover, highly crystalline CHA zeolite (Si / Al =100) with a particle size of 1.0 μm was obtained using FAU type zeolite having a Si / Al ratio of 100 as a raw material at 160 °C. for 4 days.
In recent years, the light olefin manufacturers have often confronted an unpredictable regional supply-demand imbalance between light olefins. Therefore, the olefin plant requires an olefin interconversion technology as a simple and optional process to respond quickly to the sudden imbalance of light olefins. The olefin interconversion process is typically divided into two processes: the ethylene-to-propylene (ETP) process and propylene-to-ethylene (PTE) process. The ZSM-5 zeolite was known to exhibit the highest ethylene selectivity in zeolite-catalyzed PTE reaction, and the post-modification with phosphorus could further increase the selectivity. However, the P modification was accompanied by lowering propylene conversion due to the decrease of the strong acid site, which leads to a decrease of the yield of ethylene. Herein, the post-treatment of ZSM-5 zeolite with ammonium hexafluorosilicate (AHFS) was carried out in order to improve the yield of ethylene in PTE reaction. The AHFS-treated ZSM-5 showed a considerably increased yield of ethylene than the pristine (H-Z50) and P-modified ZSM-5 (P-Z50) as shown in the figure. AHFS-treated ZSM-5 exhibits high ethylene selectivity and propylene conversion simultaneously, thus results in the improved ethylene yield. The origin of increased catalytic performance was investigated using ammonia temperature-programmed desorption (TPD) method and solid-state 27Al MAS NMR spectroscopy. From the TPD measurement, it was observed that the number of medium acid sites was increased and the number of weak acid sites was decreased. The NMR spectra indicated that the extra-framework aluminum species (EFALs), which known as having weaker acid strength than framework Al species, increased after AHFS treatment. Therefore, the AHFS treatment causes the generation of EFALs on the external surface of the zeolite and the selective increases of the medium acid site, so that thus improve ethylene selectivity and yield. The synergetic effect of Brønsted acid sites and EFALs would be suitable for acid-catalyzed PTE reaction.
Recently, in electrochemical sensing of H2O2, manganese dioxide has drawn attention as a promising electrode material due to its excellent catalytic activity which can promote H2O2 decomposition. However, many challenges are encountered with its practical application due to the poor electronic conductivity, chemical and mechanical stabilities. Considering this, in our study, we applied carbon nanowall (CNW) as the base nanoporous structure for depositing MnO2 films on it to improve the electrochemical sensor performance of MnO2.
CNWs template was synthesized by plasma enhanced chemical vapor deposition in a CO/H2 microwave discharge system. Glassy carbon with an area of 1cm2 was used as the substrate. The applied parameters of the CNWs deposition process were as follows: CO flow rate: 23sccm, H2 flow rate: 2sccm, pressure: 250Pa, microwave power: 80W, and deposition time: 60 seconds. After synthesizing CNW, MnO2 coating was anodically electrodeposited on CNWs using a three-electrode system consisting of the CNW/GC as a working electrode, an Ag/AgCl electrode as a reference electrode, and a platinum wire as a counter electrode at a constant potential of 1.0V vs. Ag/AgCl in the 0.25M Mn(CH3COO)2 solution.
The morphological and elemental characterization of the fabricated electrodes were carried out by SEM and TEM-EDX. Cyclic voltammetry and amperometric tests were applied for evaluating the sensor performance of the fabricated electrode.
In addition, new challenges of applying anodic deposition technique for synthesizing MnO2 films on porous CNW structure will be introduced and potential biosensor application will be discussed.
1. Liqiang Luo, Electrochimica Acta 77 (2012) 179– 183.
2. Chunyan Guo, Sensors and Actuators B 206 (2015) 407–414.
The utilization of surplus electricity from renewable energy resources is one of the social problems; accordingly, non-thermal plasma is drawn attention as its utilization methods.
In this study direct synthesis of liquid hydrocarbons from methane and carbon dioxide using pressure-swing plasma is studied.
In traditional Gas to Liquids (GTL) process, low-pressure conditions are preferred for the reforming reaction of methane to form syngas while high-pressure conditions are preferable for the synthesis of higher-hydrocarbons from syngas (Fischer-Tropsch Synthesis).
In addition to conventional plasma system, in this study, unprecedented pressure-swing of CH4-CO2 plasma is applied for the solution of trade-off pressure conditions.
It is expected that dissociative reaction by plasma and synthesis reaction by pressure-swing will proceed concertedly and liquid hydrocarbon will be synthesized directly.
As pressure-swing plasma system, we utilized a uniquely modified diaphragm pump as a pressure-swing reactor with creeping discharge which develops inside the chamber of reactor.
Electrode has structure that quartz plate as dielectric put between cupper tape as ground electrode and carbon tape as high-voltage electrode and plasma forms near ground electrode. We can also put other metal (e.g. Co foil, Fe foil, and so on) on cupper tape.
Pressure-swing range is approximately 1 – 3 bar, frequency of swing is ~25 Hz. Typical applied voltage of plasma is 9 kV, power consumption of plasma is approximately 4.5 W. Timing of plasma discharge is pulsed and manipulated by micro-controller, typical pulsed frequency is 100 Hz.
The conversion of methane and carbon dioxide and the selectivity of product hydrocarbon are investigated by GC.
From the experiments, we observed that the conversion is improved and the selectivity of product hydrocarbons is shifted to heavier hydrocarbons in pressure-swing condition.
Moreover, we discussed and considered the relation between discharge timing and the selectivity.
Lithium air battery is one of the most promising energy storages due to its high theoretical energy conversion comparing to conventional lithium ion battery. However, development of lithium air battery to practically use in actual application has been faced many problems such as dendrite on lithium anode, decomposition of electrolyte and pore clogging on cathode. Focusing on cathode, many researches applied highly porous and functional carbon materials such as graphene or carbon nanotube to fabricate cathode by evaporation method. Despite the good properties, those materials too expensive to practically use. Moreover, evaporation causes aggregation of carbon material due to high surface tension, decrease surface area for cathodic reaction.
In this work, we applied carbon black (CB), which is extremely cheap material comparing to graphene or carbon nanotube, as the active material for cathodic reaction and Polyvinylidene fluoride (PVDF) as the binder. Moreover, [C4mim][Tf2N] ionic liquid was impregnated in PVDF to improve ionic conductivity, electrical conductivity and reaction gases selectivity. Those explained materials were dispersed in N-Methyl-2-pyrrolidone (NMP) and dried by supercritical carbon dioxide at 20 MPa, 40 °C. Cathodes were assembled into meshed coin cell with 1 M LiTFSI/TEGDME electrolyte. The capacity was tested with air, pure O2, O2/CO2 mixing gases at 0.075 mA cm-2.
CB/PVDF cathode dried by supercritical CO2 has 92% of porosity, while CB/PVDF/[C4mim][Tf2N] cathode has porosity around 84%. Despite the lower porosity, Li-air battery applied CB/PVDF/[C4mim][Tf2N] cathode provides capacity at 23.2 mAh cm-2 (42.4% efficiency), which is larger than CB/PVDF cathode at 19.6 mAh cm-2 (35.7% efficiency). The discharge products were confirmed as Li2O2 by XRD in both cases. The improvement of specific capacity is considered that ionic liquid gel enhances ionic conductivity and improves selectivity and solubility of reaction gas on surface of cathode.
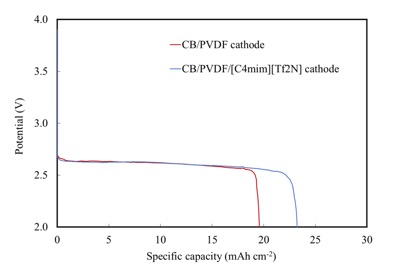
Bi2Te3 type semiconductor is well known as a thermoelectric conversion material generally used in a temperature range around room temperature, and is also put to practical use as a Peltier element for cooling. The Bi2Te3 type semiconductor is generally produced from depositing fine metal powder by a melt under high vacuum or inert atmosphere, and sintering from a ground alloy or mechanical alloying method, but these synthesis process are complicated and the cost benefits are low. Therefore, direct synthesis of single phase fine particles was attempted by preparing a neutralized co-precipitate from each metal chloride mixed aqueous solution, adding a reducing agent and subjecting it to hydrothermal treatment. An aqueous solution of chloride of Bi, Te, Sb, and/or Se was mixed in an appropriate ratio, and the aqueous solution of NaOH was added dropwise with stirring vigorously to prepare a co-precipitate of each composition. HCOOH or NaBH4 as a reducing agent is added to the aqueous solution, hydrothermal treatment is performed at 473 K. for 24 h in an autoclave equipped with a Teflon bottle, the product is filtered off with suction with a 0.45 μm filter, and after distilled water washing, and dried at 333 K or less. As a product, a plate-like fine crystal having a diameter of about 100 nm and a thickness of about 20 nm was obtained, in which the chemical composition was almost uniform in all of Bi2Te3, (Bi, Sb) 2Te3 and Bi2 (Se, Te) 3 ). The electrical resistance and Hall effect for representative product samples were measured. As the results, both products show relatively low efficiency, and it is found that Bi2Te3 and Bi2 (Se, Te) 3 show n-type, and (Bi, Sb) 2Te3 shows p-type semiconductors.

Examination of fouling mechanism in heat exchanger is demanded to minimize fouling trouble. This study analyzed fouling deposits recovered from heat exchanger to reveal the chemical structure of these deposits. The basic structure of the deposits was determined by nuclear magnetic resonance, pyrolysis and X-ray diffraction. The deposits consisted of diene and iron oxide. The CHNS elemental analysis showed that H/C atomic ratio was approximately 1.4 and the deposits had oxygen. The infrared spectroscopy analysis indicated that the deposits had carbonyl groups.
Acknowledgment: This study was supported by the Advanced Production System Project Committee of the Society of Chemical Engineers, Japan.
Introduction
Berry fruits are well known to contain large amounts of polyphenol compounds. Among them, flavan-3-ol and anthocyanidin derivatives are a group of secondary metabolism compounds currently attracting a great deal of attention owing to their health benefits. Berry fruits are often consumed as processed products such as juices and jams in addition to fresh fruits. Most processing methods are heat-drying, but there is little detailed research on how polyphenol compounds contained in fruits change as a result of processing. Therefore, we wanted to chemically track how the polyphenol compounds contained in the fruit change with processing. Therefore, we selected Red Ruspberry (Rubus idaeus L.) as an experimental model plant because it is widely known for edible fruits and is rich in useful polyphenols such as flavan-3-ol.
Results and discussion
The HPLC chromatogram of the fresh raspberry-MeOH extract is shown in Figure 1. The peaks in the chromatogram were identified as catechin (1), epicatechin (2), procyanidin B4 (3), procyanidin trimer (4) and hydrolyzable tannin (5), respectively. In dried fruits, it was confirmed that hydrolyzable tannin was decreased. Measurement of DPPH radical scavenging activity, which is an indicator of antioxidant activity, showed that the activity was reduced in dried fruits. In this report, we will discuss in detail the chemical changes of polyphenol components by drying and heating.
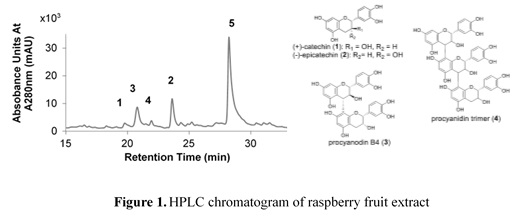
Reactive dyes, which are most widely used for dyeing cotton, have reactors capable of chemical reaction with the hydroxyl groups of cotton fiber. Depending on the type of reactor, the dyes vary greatly in the size of the reactivity and the dyeing temperature. Therefore, the optimum dyeing temperature depends on the reactor.
The dye molecules adsorbed by the addition of the initial neutral salts are markedly less substantivity between the fibers and the dye when the alkali is added. At this time, dye molecules and fibers are covalently bonded by chemical reaction.
The alkali catalyzes the reaction when the dye and the fiber react with each other because the concentration of the OH group in the fiber is increased by the addition of the alkali to increase the reaction. At the same time, however, the OH group concentration of water is also increased, facilitating the hydrolysis, and the pH of the bath is determined by the kind and amount of alkali. The reaction proceeds rapidly as the pH of the bath is high.
In this study, we used the regression analysis to determine the optimum alkali injection timing at the time of the exhaustion equilibrium. The point in time when the substantivity between the fiber and the dye was significantly lowered was calculated and it was decided that the optimum alkali injection time point.
The main factors affecting the primary exhaustion equilibrium during the dyeing process with reactive dyes are neutral salts, that is, in general, the neutral salt, which inhibits the electrostatic barrier(Donnan potential) between fibers and dyes, Eliminate the repulsive force on the fibers.
It also facilitates the action of the Van der Waals force, facilitating the access of the dye to the fiber surface, thereby enhancing the adsorption of the dye.
The amount of neutral salt used is determined by the amount of dye. The more dyed dyes require more neutral salts, the more the amount of neutral salts added, the higher the adsorption of dye in the dyes. This is the basic condition for the next fixing reaction step. In order to increase the adsorption of the reactive dye which is lower than the direct dye, the use of a large amount of neutral salt is required, but the adsorption rate is not improved.
In this study, the basic method of determining the optimum injection timing of alkali additives was analyzed by analyzing the input method and the input amount of the manganese which are the main factors of the dyeing equilibrium of reactive dyes.
Poly(N-Isopropylacrylamide)(PNIPAM), a thermo-sensitive polymer, is well known for undergoing coil-to-globule transition at lower critical solution temperature (LCST) of 32 °C. As this LCST is close to the temperature of the human body, considerable research efforts have been directed towards its potential use in drug delivery systems, functional materials, and humectant absorbents. Conventionally, PNIPAM is produced in aqueous system through the addition of an initiator, however these methods require continuous stirring and lengthy treatment times (more than 6 h). Recently, plasma initiated polymerization methods have been attracting attention because of the generation of free radicals in the process. To generate plasma, pulsed power is commonly used. It is an instantaneous form of energy that, when temporally compressed, can produce a tremendous amount of electricity. This study presents a novel, more efficient PNIPAM polymerization method that uses pulsed arc discharge. An optimal raw material conversion, over 90%, is obtained with an initiator concentration of 0.1mol/L at 4 pps for 1 min in the presence of 2,2´Azobis(2-Methylpropionamidine) Dihydrochloride as an initiator. Furthermore, the effect of the initiator concentration and the distance between the cathode and the aqueous solution are studied. When the cathode is submerged in the aqueous solution, these conditions produce a yield over 80% and an average molecular weight of 261±2.61 (see Table 1).
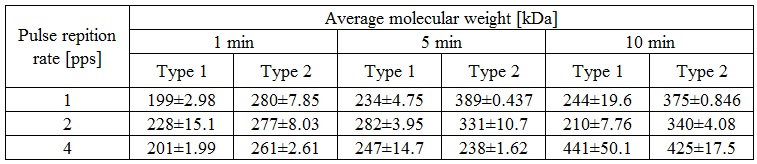
Plasma technology has great potential for converting excess renewable energy into value-added chemicals. In the previous research in our laboratory, we were able to synthesize ammonia efficiently by pressure swing of N2-H2 plasma. In this research, we will consider the process of synthesizing ammonia by pressure swing of N2-H2O plasma.
Nitrogen gas and water vapor were used as feed gas, electrodes were inserted inside the pressure swing reactor to generate plasma, and then the product gas was analyzed. Nitrogen gas was bubbled into pure water and water vapor, which is a hydrogen source of ammonia, was introduced. The pressure-swing was performed by using a modified diaphragm pump. The product gas was trapped by bubbling into pure water, and the obtained aqueous solution was analyzed by HPLC.
The amount of ammonia synthetic efficiency when the reaction is carried out under a constant pressure and under pressure swing is shown in Figure. By performing pressure swing, the efficiency of ammonia synthesis increased from under a constant pressure.
Moreover, when the metal electrode was changed from copper to platinum, an increase in the synthesis efficiency of ammonia was confirmed under both constant pressure and swing conditions. This indicates that the rate-determining step of ammonia synthesis in this study is a surface reaction, and pressure swing enhances the surface reaction.
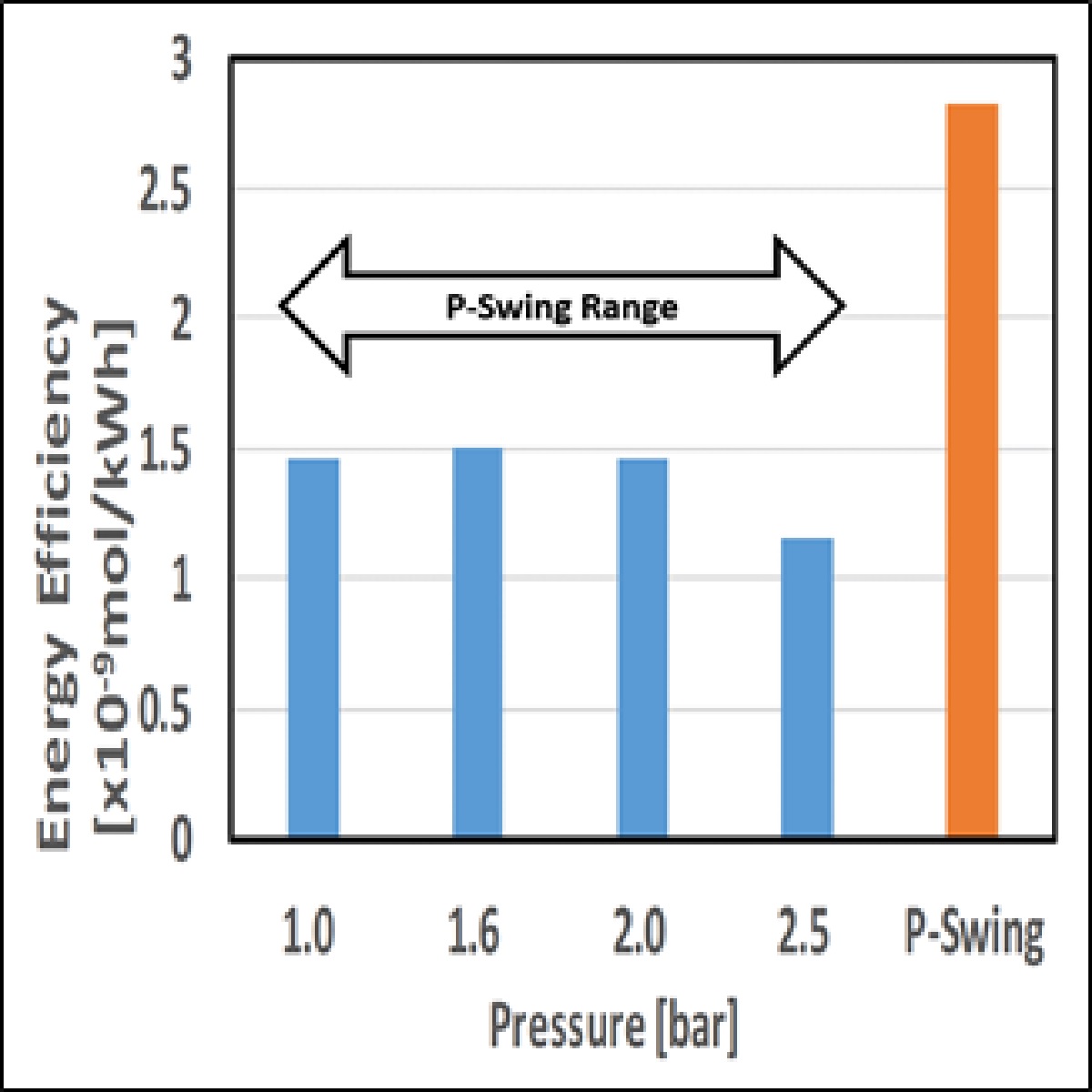
It has been well known that Ammonia is a very important chemical substance for the life of humankind. Simultaneously, conventional ammonia production process needs nitrogen gas and also hydrogen gas which has been produced from either LNG or coal. This study is regarding ammonia production from nitrogen gas and water (Do not hydrogen) by plasma/liquid interface (P/L reaction). We have developed the unique reaction which produces ammonia at the plasma gas phase/liquid (water) phase interface (P/L reaction locus). Although the outermost of surface water molecules on the water phase show a unique orientation, the hydrogen (H) of water molecule on a water surface is possibly oriented toward the gas-phase side. Activated nitrogen extracts H from H2O at water phase surface to produce ammonia, which dissolves in the water.
In excited nitrogen gas (N2 plasma) phase, several excited state have been reported. These include atomic nitrogen (Natom), excited nitrogen molecules (N2*), nitrogen ions (N2+), and the like, which have different lifetimes and reactivities. We have already shown that atomic nitrogen is highly reactive by quantitative analysis of N(4S) nitrogen atoms. Furthermore, it has been reported that increased the amount of ammonia production starting from low reactivity nitrogen species (N2* and N2+) pathways by activating the water phase. Upon UV irradiation onto the water phase, the activated nitrogen with low reactivity also promotes a reduction reaction system using water molecules.
In this study, we examined the P/L reaction pathways that changed the conditions of the water phase. The reaction pathway of ammonia production was examined by changing the pH, changing ionic strength, and changing the amount of water.
This poster focuses on the utilization of the synergy of microwave irradiation and graphene-based catalysis as applied to various reactions related to biomass conversion processes including cellulose depolymerization, glucose conversion into platform chemicals, and transesterification/esterification/etherification of oil fractions and products in biomass. The result are part of the research contributions of the Japan team to the recently concluded JST e-ASIA Joint Research Program on “Development of Functional Nanocarbon-Based Catalysts for Biomass Conversion Processes.”
Conversion and utilization of inedible lignocellulosic biomass, which mainly consists of cellulose, hemicellulose and lignin, have greatly attracted attention to reduce carbon dioxide emission and to establish the sustainable society. Cellulose and hemicellulose are polysaccharides, which can be converted into valuable products and fuels. Lignin is a three-dimensional polymer of aromatic compounds with cross-linking via C-O-C ether bonds and C-C bonds, which can be converted into aromatic products by depolymerization; however, the lignin conversion to aromatic monomers is still challenging. We reported the bond cleavage reactions between aromatic monomers using lignin model compounds in supercritical water at 673 K in the presence of supported palladium, platinum, and rhodium catalysts without added hydrogen gas and without causing hydrogenation of the aromatic rings in the previous paper. Thus, we applied this technique to the lignin into aromatic compounds. In this paper, we found that lignin could be converted into aromatic monomers in supercritical water at 673 K over supported metal catalysts. We investigated the effect of supported metal catalysts on lignin deporimarization into aromatic monomer in supercritical water.
Acid-catalyzed condensation between alcohols and carboxylic acids to produce ester compounds (i.e., Fischer esterification) plays an important role in chemical industries. Since Fischer esterification is an equilibrium reaction, it is mandatory to shift the equilibrium to obtain the ester products with high yields. Increasing reaction temperatures more than 100 °C to remove water by-product from reaction systems is a typical procedure to shift the equilibrium. However, undesirable side reactions might occur at higher temperatures (e.g., degradation of reactants/products, etherification of alcohols, and condensation between alcohols and acid catalysts). In addition, separation/purification of the products generally requires distillation process, as well as neutralization process of acid catalysts, which severely raises energy costs.
In the present study, we have prepared ionic liquid catalysts (ILCs) that undergo phase separation with ester products, while water by-product is selectively absorbed in the ILC phases [1,2]. The phase separation of esters and water efficiently shifted the equilibrium at moderate temperature, and high conversion values over 90% was obtained for the synthesis of wax esters from long-chain fatty acids/alcohols. The ILCs were not contaminated in the ester phases. On the other hand, the partition of unreacted acids/alcohols and ester products was strongly dependent on the structure of ILCs. Details on the relation between the partition behavior and reactivity will be discussed. Moreover, our preliminary studies to utilize flow reactors will also be presented.
References
[1] Y. Kohno, T. Makino, M. Kanakubo, React. Chem. Eng., 2019, 4, 627-633.
[2] Y. Kohno, T. Makino, M. Kanakubo, Fluid Phase Equilib., 2019, 490, 107-113.
When ultrasound is irradiated to water, degassing, that is, decrease of dissolved gas concentration occurs [1]. However, the effect of ultrasonic frequency on degassing behavior is few reported. In this study, the change in dissolved oxygen with ultrasonic irradiation time was investigated for a wide range of ultrasonic frequencies.
Experiments were carried out using a cylindrical vessel with a 56 mm in inside diameter. Transducers (Honda Electronics) attached to the vessel were a Langevin type multi-frequency transducer with 45 mm in diameter at frequencies of 22, 43, and 129 kHz, and disk type transducers with 50 mm in diameter at 209, 308, 400, 514, 1018, and 1960 kHz. Transducers were driven by a power amplifier. Ultrasound at 15 W of the ultrasonic power was directly irradiated to a 100 mL sample in the vessel. The ultrasonic power which was the energy applied to the sample per unit time was obtained by calorimetry. Ultrapure water was used as the sample. The degassing behavior was measured by the fluorescence dissolved oxygen meter.
The effect of ultrasonic frequency on time change of the dissolved oxygen by ultrasonic irradiation is shown in the figure. The dissolved oxygen decreases with ultrasonic irradiation time, and the decrease rate is maximum at 300 kHz. Dependence of sonochemical reaction on the ultrasonic frequency at 15 W was investigated by the KI method. In this method, triiodide ion is generated from iodine ion by ultrasonic cavitation. Sonochemical efficiency was also maximum at 300 kHz. Because a lot of cavitation bubbles generated in the sample at 300 kHz, the dissolved oxygen decreased significantly. It was proved that frequency dependences of sonochemical efficiency and degassing rate correlate each other.
[1] N. Gondrexon, V. Renaudin, P. Boldo, Y. Gonthier, C. Pettier, Chem. Eng. J., 66 (1997) 21-26.
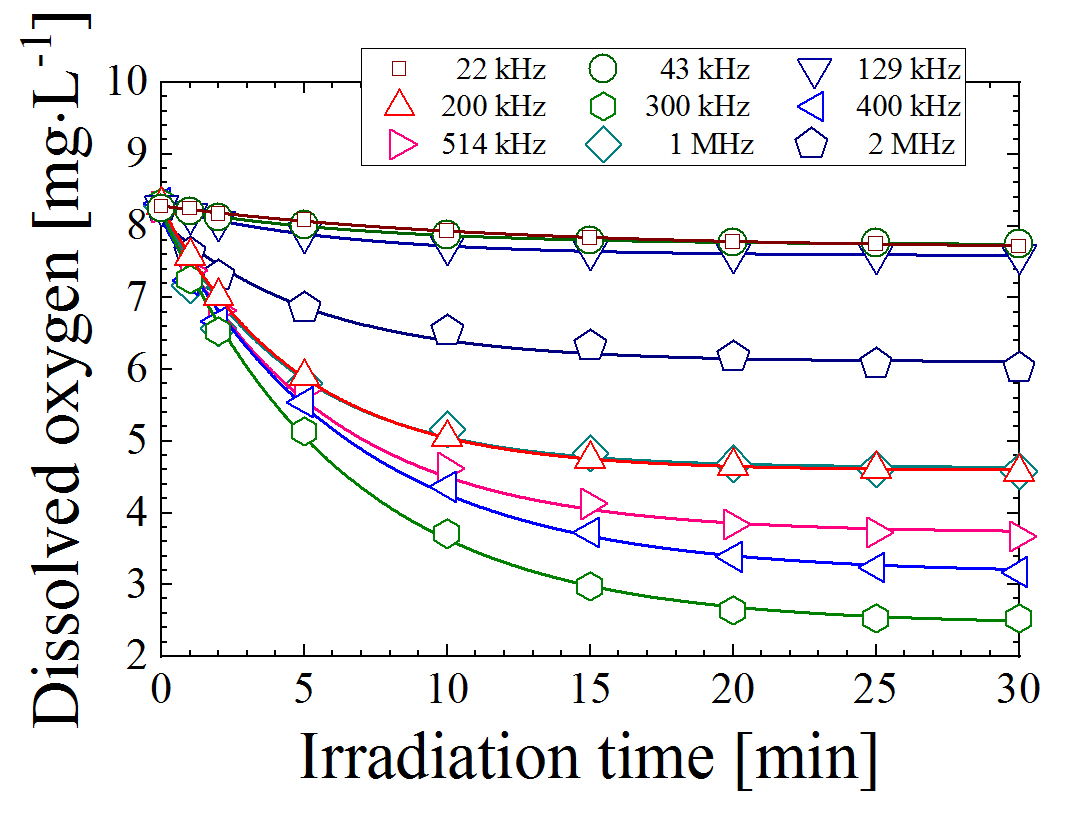
Recently, attention about phenomena and effects caused by supply of fine bubbles to aqueous solution is increasing. With regard to fine bubble species, the dissolution phenomena of hydrogen has not been clarified. Reduction of the concentration of oxygen active species owing to the dissolution of hydrogen fine bubbles is expected for application of food, substance synthesis, and environmental conservation fields. In this study, effects of molar fraction and bubble size of hydrogen/nitrogen fine bubbles on dissolution rate, solubility and reaction characteristics with oxygen species active species were investigated.
We have previously reported that OH-radical formation was enhanced by the existence of microbubbles under the specific frequency (45 kHz) of ultrasound irradiation. In the present work we have performed the experiment of ultrasonic degradation of azo dyes with various frequencies with and without microbubbles. It was found that for 45 kHz ultrasonic irradiation microbubbles enhances the degradation of azo dyes. To confirm that the effect was due to the cavitation bubbles formed by the irradiation ultrasound, we have introduced microbubbles with various gases and it was found that Kr and Ar gases have much larger formation of OH radicals, while He gas was very few effect on the enhancement of OH radicals. It was concluded that the cavitation bubbles were formed due to the ultrasonic irradiation and OH radicals were formed from these cavitation bubbles. Thus microbubbles have some effects on the enhancement of cavitation bubbles only in the case of 45 kHz ultrasonic irradiation.
In recent years, a great interest has been taken in microwave enhanced chemical reaction and many research have been conducted using microwave as a heat source. It is said that microwave heating has many advantages such as generating a uniform temperature distribution within a irradiated medium, quick heating, enhancing a reaction rate and improvement of selectivity on reaction products.
On the other hand, ultrasonic are also utilized for chemical reaction and it is reffered to as sonochemistry. There arises some effects by ultrasonic such as macroscale and microscale mixing due to the moving or oscillation of bubbles generated by cavitation and hot or high pressure spot derived from the collapse of bubbles.
In this study, experiments of esterification were carried out where irradiation intensity of microwave and ultrasonic were changed, and the effect of both irradiation on the conversion or reaction rate of esterification was investigated. The irradiation of microwave and/or ultrasonic was conducted until the reagent temperature reaches 333K.
As a result, when the irradiation of microwave and ultrasonic were introduced simultaneously where microwave power was varied with constant ultrasonic power, apparent reaction rate constant became larger than in the case that only microwave was irradiated. As for conversion, which is the result determined by reaction rate and reaction time, it decreased as microwave power become larger under constant ultrasonic power since the reaction time shortened because of quick ascent of temperature due to large microwave power , whereas it increased as ultrasonic power become larger under constant microwave power.
In this work, a spiral microchannel reactor was fabricated, and then applied to a selective synthesis of monoglycerides (MON) from palm fatty acid distillate (PFAD) and glycerol via esterification in the presence of sulfuric acid. An angle-less stainless-steel microchannel reactor with 200-cm length and 1×1 mm cross-sectional square-shape channel was designed to overcome the pressure drop related to a high viscosity of PFAD and glycerol. The flow patterns of both reactants were determined by using different fluid mechanic equations. The effects of reaction temperature and residence time on the PFAD conversion and the MON selectivity were investigated. Moreover, the results were compared with those obtained from stirred-tank batch reactor. It was found that the esterification of PFAD with glycerol under batch conditions gave the highest PFAD conversion (>95 wt.%) but the MON selectivity was only 40% due to the formation of di- and tri-glycerides. At a similar conversion, the spiral microchannel reactor gave higher MON selectivity than the batch reactor. The suitable condition for MON synthesis in the spiral microchannel reactor was 2 wt% sulfuric acid (based on the PFAD weight) at 200 °C and 2 h residence time under which the PFAD conversion and the MON selectivity were 81 and 74 %. The selective MON formation was explained by counter rotating vortices flow and solubility parameter of reactants and products.
To enhance the thermal controllability of the reaction system, a compact reactor was introduced, where heat transfer with both cooling media and cool feed gas was utilized. A computational fluid dynamics model was developed to analyze detailed profiles of mass, momentum and heat, and it was shown that the compact reactor could efficiently remove the generated heat. The space velocity and feed temperature were manipulated to increase the production rate, and the proposed reactor could produce twice of methanol with its peak temperature maintained below that of the conventional reactor. When the diameter of the catalytic bed in the compact reactor was increased, the reaction stayed in the kinetic regime, resulting in high methanol productivity as well as the maximum utilization of the bed.
Extraction with slug flow (alternating flow of different phases) gives fast mass transfer rates and rapid phase separation than the conventional extraction operations. Therefore, there are many advantages such as a reduction in volume of the device and a drastic decrease of the solvent used in the process. The circulating flow in each liquid segment and the interface area per the unit liquid volume largely depend on the geometry of the liquid segment. When the extraction by the slug flow is performed in a narrow diameter tube, the pulse flow (pulsation) of the pump that supplies the liquid gives a great influence on the flow condition in the tube. When the movement of each liquid segment is not kept uniform, the liquid flow disturbance cannot ignore the influence on the extraction rate. So, this unstable change of the flow is not desirable in analysis of extraction rate and device design. Therefore, a syringe pump with little pulsation has been used in many studies on the slug flow. The syringe pump is suitable for extraction by the slug flow since it shows no pulsation. However, it is not possible to supply continuously more liquid than the volume filled of the syringe. This indicates that it is not difficult to handle more than the volume of the syringe. The development of a stable flow liquid pump that supplies liquid without pulsation is expected.
In this study, two diaphragm pumps were interlocked to control the flow rate and the reciprocation frequency of the membrane. We succeeded in generating a liquid-liquid slug flow with a fixed length of about 1-6 mm. It was also confirmed that the generated slug flow was a stable flow of about 1 hour. We have found the possibility of becoming a new slug flow generation device.
Extraction by slug flow gives high mass transfer rate and rapid phase separation than the conventional extraction operation. Therefore, there are many advantages such as the volume of the device becomes small and the solvent drastically reduced. The slug flow extraction is expected as a sophisticated separation process. Especially, these advantages become more prominent in the multistage extraction. In this study, the extraction by the slug flow was applied to mutual separation of lithium, cobalt and nickel ions in the mixed aqueous solution. By combining forward and backward extractions, cobalt ions could be recovered with concentrating about 1000 times. We succeeded in developing a process with lithium, cobalt, nickel ion purity of 85% or more and recovery rate of 90-95%. Furthermore, it was also confirmed that the flow state of the liquid is influenced by the hydrophobicity (PTFE tube) and hydrophilicity (glass tube) of the extraction tube. The overall mass transfer volumetric coefficient is affected by (1) the interface area of the oil phase and aqueous phase, (2) the strength of the circulating flow in each segment of the liquid, (3) whether the mass transfer resistance is in the aqueous phase or the oil phase, and (4) the volume flow ratio of the oil phase and the aqueous phase, and so on. In general, the total mass transfer volumetric coefficient increased when the liquid with low mass transfer resistance wetted the wall of the extraction tube. This suggests that the wetting of the pipe has a great influence on the extraction rate in the slug flow extraction. It is necessary to select the tube material for operation properly. Furthermore, we confirmed that the overall mass transfer volumetric coefficient can be easily calculated from the experiments and the extraction rate can be simulated by a numerical method.
Quenching flow method using flow microreactors is known to be suitable for kinetic analysis of chemical reactions. This is because the characteristics of the flow microreactors such as fast mixing, precise residence time control, and precise temperature control makes it possible to control precise reaction conditions. The product can be accurately analyzed by various analytical methods such as gas chromatography. We have already reported kinetic analysis of organolithium reaction by quenching flow method using flow microreactors. Herein, we apply this method to the Schotten-Baumann reaction, which is the reaction of amine and acid chloride in the presence of basic aqueous solution. In this presentation, we report the kinetic analysis of the amidation and the comparison of the kinetic difference between various amines.
Among multiphase reactors in the chemical and pharmaceutical industries, packed bed compact reactors with internal diameters on the order of millimeters or micrometers are of interest. However, the design and operation method of packed bed compact reactors has not been established because it is difficult to analyze the flow pattern, which greatly influences heat and mass transfer performance, by visualization. In this study, an experimental system consisting of a packed bed compact reactor was constructed, and a method of estimating the flow pattern in the reactor was developed through numerical simulation as well as experimental analysis of the measurement results of voltage or differential pressure. The results show that when gas and liquid were supplied to the reactor, the developed method made it possible to distinguish the flow patterns of trickle flow and pulse flow from the shapes of the measured voltage or differential pressure and to estimate the pulse frequency of pulse flow (i.e., peak frequency of power spectral density), which is useful in grasping the degree of mixing of gas and liquid. It was suggested that the design of the mixer capable of producing shorter gas and liquid slug as well as the design of the reactor inlet should be developed in order to realize the process with higher gas and liquid mixing.
Introduction
Recent medical/orthodontic curable compositions have acquired high physical properties by using the newly designed long-chain silane coupling agent[1]. However, since the synthesis process required hydrosilylation as shown in Figure 1, it was essential to remove the platinum group complex from the synthesized material. In this study, we synthesized a long-chain silane coupling agent from a both-terminal-equivalent compound[2] such as ethylene glycol as a substrate using a micro flow reactor, and evaluated the influence of various synthesis conditions on the yields, etc. of the resultant silane coupling agent.
Results and Discussion
In comparison to the batch method, the flow method more efficiently synthesized mono-substituted products. Since the mixed solutions of 2-isocyanatoethyl methacrylate and ethylene glycol were heterogeneous, the reactions may have occurred at the liquid-liquid interface. The flow method provided larger surface area of the liquid-liquid interface than the batch method because the flow method produced fine emulsions using a chip (static mixer) for mixing the solutions. This resulted in more efficient reactions compared to the batch method.
Reference
[1] Fuchigami K, Fujimura H, Teramae M, Nakatsuka T. Precision synthesis of a long-chain silane coupling agent using micro flow reactors and its application in dentistry. Journal of Encapsulation and Adsorption Sciences, 2016, 6(1), 35-46.
[2] Huang X, Liu Y, Wang S, Zhou S, Zhu D. Synthesis and self-assembly of 2,9,16-tri(tert-butyl)-23-(10-mercaptodecyloxy)phthalocyanine and the application of its self-assembled monolayers in organic light-emitting diodes. Chemistry - A European Journal, 2002, 8(18), 4179-4184.
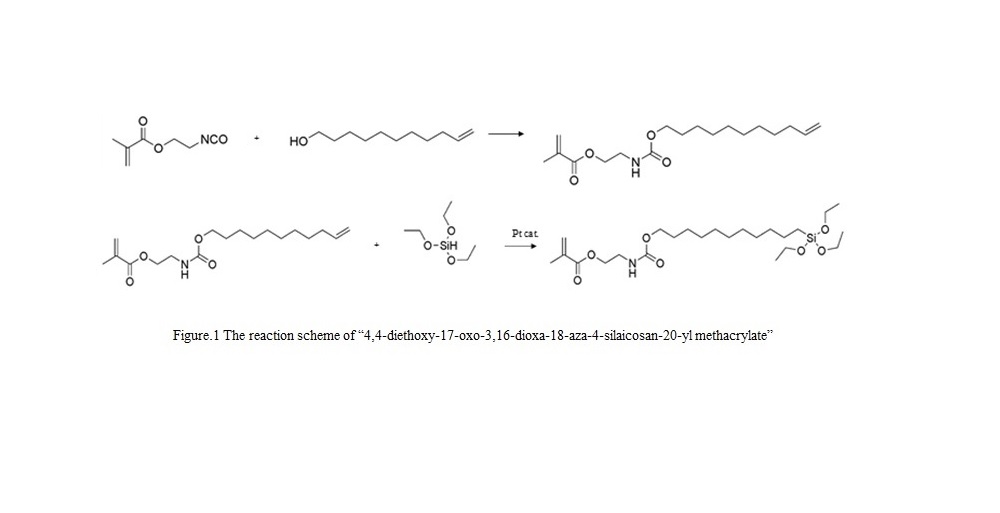
The main objective of this study is to investigate the flow pattern with gel reaction inside a continuous circular flow pipe and to identify the governing dimensionless number of flow patterns with gel reaction. The flow channel consists of a transparent main flow pipe and a transparent branch flow pipe which is connected to the main flow pipe. 10 mass% PVA and 3 mass% borax solutions are used as working fluids. 10 mass% PVA solution flows in the main flow pipe with steady-state conditions. Reynolds number (Rem) based on the mean velocity and the diameter of the main flow pipe is less than 30. The borax solution are injected from the branch flow pipe to the main flow pipe. The duration of the injection time and injection volume flow rate from branch flow are varied correspondingly to deliver a fixed volume of borax solution. At first, three flow patterns are observed inside of flow pipe, Capsule (C), Capsule + Stretched (CS) and Capsule + Stretched + Fingering (CSF) flow pattern. To identify the dimensionless number of three flow patterns with gel reaction quantitatively, we introduced Rem and β which is the volume flow ratio of the main flow to that of the branch flow. The results show that when β is less than unity, C flow forms and β is the main parameter that determines the flow pattern with gel reaction. When β exceeds unity, Rem is the main parameter that determines the flow pattern with gel reaction. When Rem exceeds 10.0, CSF flow forms. These results indicate that β and Rem play key dimensionless number in the gel formation and flow stability in a flow channel.
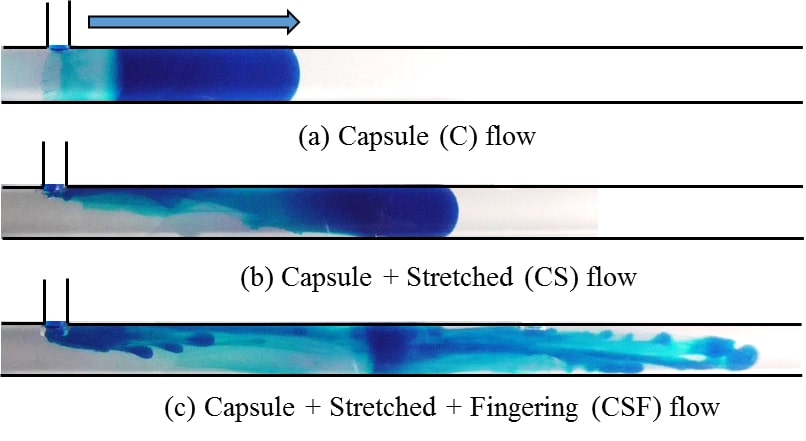
Hydrogenation is an important chemical reaction process in the production of petrochemical products, perfumes, and pharmaceutical intermediates. A gas-liquid-solid three phase reaction was carried out in a slug flow reactor in order to improve the apparent reaction rate by enhancing mass transfer rate of gas to the solid catalyst surface through the liquid. Hydrogenation of alpha-methyl styrene (AMS) using hydrogen gas was employed as the model three-phase reaction. The reactor was an aluminum tube of length 300 mm and inner diameter 4 mm. A thin alumina layer was formed on the inner wall and contained palladium as a catalyst. The hydrogen and AMS were introduced into a T-joint installed at the upstream of the reactor to form the gas-liquid slug flow. The AMS conversion obtained by the slug flow was much higher than that without slug flow. In order to examine the effect of the hydrogen absorption promotion by the circulating flow in the AMS slug, the total flow rate of the reactants was varied while the lengths of the gas and liquid slug were fixed to be constant. Although the absorption must be increased due to the faster surface renewal, only slight improvement could be observed. When the total flow rate was fixed and the ratio of the gas slug length to the liquid slug length was increased, the apparent reaction rate was improved significantly. These results indicate that the diffusion in the thin liquid film directly toward the solid catalyst was dominant for the reaction rate enhancement. Furthermore, the influence of the specific surface area of the liquid film layer was investigated from the mass transfer model by conducting the reaction experiments using the tubular reactors with different inner diameters.
In recent years, with the increase of global energy demand and adaptation to a sustainable society, there is a demand for higher efficiency of energy use in chemical plants. As one of the methods to improve the efficiency and energy saving of the chemical reaction process, this study examined the electric heating catalytic reaction process. The electric heating is a system in which Joule heat is supplied to a catalyst carrier having electric resistance. In this system, thermal energy is directly supplied to the reaction site. The etched aluminum foil was used for a catalyst carrier. This material has through holes at a density of about 10,000 holes / mm2 and its diameter can be controlled to 1 to 4 μm. A reaction space such as a microreactor can be realized by using the through holes as reaction channels. Further, the catalyst metal can be supported by making the wall surface of the flow path porous, and it also has a function as a structured catalyst. With regard to electric heating, thermal energy is generated by supplying current to the base aluminum. In the evaluation of the electric heating performance, the model reaction was the methanol steam reforming. Evaluation factors were temperature rising rate, temperature reached, power consumption, and hydrogen generation rate. As a result of conducting the activity test, energy consumption was reduced by 40% in the electric heating as compared with the power consumption of the process by the external heating. In the future, we will study the temperature non-stationary operation in order to evaluate the startup performance of electric heating.
Multiphase reaction systems for organic synthesis are encountered in many chemical industries producing petrochemical derivatives, fine chemicals and pharmaceuticals.
Operating multiphase flows in the microscale has proved to be effective owing to its enhanced mass transfer. Additionally, microreactors bring the advantage of safety, easy control and flexible scalability by numbering up compared to the large chemical reactors. In microstructured devices where the diameter falls into the range of 100 to 1000 microns, time scales are small and molecular diffusion is the dominant driving force for mass and heat transfer. Enhancing the performance of microreactors requires improving advective mass and heat transport which can be realized by implementing helical structures. The flow in the helical structures is known to generate secondary flow vortices in the radial and tangential directions, which are believed to enhance the convective transport.
In order to provide an insight into the transport phenomena taking place, this research work aims to simulate multiphase flows with a focus on the effect of helical structures on mass transfer. A new numerical model is suggested to reduce the computational domain from a full-scale helical structure to one straight 3D slug. The numerical model is built using the Volume of Fluid (VOF) method and simulations are run using the open source OpenFOAM software. A multiphase simulation of water-air segmented flow showed that the diffusion of oxygen from air to water is faster in a helically coiled microcapillary compared to a straight microcapillary even at low Reynolds numbers.
Mixing characteristics under laminar flow conditions was examined. A thin tube is co-axially inserted into the channel through which the solution of reactant A flows. The solution of reactant B was co-currently fed from the thin tube into the A solution. The mixing characteristics of B into A in the channel were evaluated under the condition of flow ratio A : B = 100 : 1. An orifice was installed downstream of the junction of the two solutions for promoting mixing. The influence of the flow rate ratio of the two solutions, the installation position and the geometrical shape of an orifice on the mixing characteristics was evaluated by Dushman reaction. As a result, in the co-axial double tube type mixer, the mixing characteristics of the two solutions became better as the total flow rate increased, even under co-current and laminar flow conditions. Moreover, it was found that the double tube type mixer was much better compared with mixing characteristics in a beaker stirred by a magnetic stirrer. The mixing characteristics when the orifice was placed downstream of the confluence point were significantly better than those without the orifice under the same Reynolds number condition. These results are considered to be due to the promotion of mass transfer in the greatly stretched thinner flow from the inner thin tube because the flow rate of the solution in the annular region is greater than that from the inner thin tube. When the orifice is inserted, it is thought that the contraction flow when passing the orifice and the expansion from there have developed the same effect. These flow characteristics of the flow from the inner thin tube were also confirmed by numerical simulation.
We describe the stability of liquid-liquid slug flow in this study. Knowledge of slug stability is necessary to utilize the liquid-liquid slug flow for unit operation. Fragmentation or coalescence of slugs causes unexpected reaction performance and extraction efficiency, and affect the productivity or product purity. Establishment of the stable operation guideline for the liquid-liquid slug flow is necessary.
We made the liquid-liquid slug flow by using dodecane as an organic phase and water colored with rhodamine B as an aqueous phase. The stability of slug flow based on two control systems was examined: active slug formation with solenoid valves and passive slug formation without solenoid valves. The optical sensor was set in the flow path to measure the slug velocity. The experiment was conducted with different aqueous slug lengths.
We depicted the map for the stable slug operation, in order to the capillary number and valve frequency. Then, the behavior of slugs at various flow rates and slug lengths was investigated. When aqueous slugs of different lengths were alternately formed, a long-length slug caught up with a short-length slug. Optical measurement suggested that the velocity of the aqueous slugs was faster than the flow velocity assuming the plug-flow, and the long-length slug flowed faster than the short-length slug.
In conclusion, the stability of the liquid-liquid slug flow depended on the capillary number and the valve frequency. The uniform length of the dispersed slug contributed to the steady operation.
Hydrogen obtained by water electrolysis using electric power derived from renewable energy is attracting attention as a clean next-generation energy. The difficulty of storage and transportation of hydrogen can be solved by converting hydrogen to chemical hydride. The production of chemical hydride with hydrogen derived from water is important technology for hydrogen energy society. The methylcyclohexane (MCH) is one of useful hydrogen carrier due to about 6 wt% of its high hydrogen content and low melting point. The system combining hydrogen formation by electrolysis of water and hydrogenation of toluene to form MCH will be useful to produce chemical hydride from water. The hydrogenation process can be expected as a new energy storage method in a decentralized society because the device can be miniaturized while using electric power derived from individual renewable energy. Hydrogenation of toluene to produce MCH proceeds in the presence of catalytic Pt/Al2O3 powder at the permeation side of Pd-Ag membrane electrode in our previous study. In this case, it is difficult to fill up a lot of catalyst powder in small space. Structured catalyst will solve the structured problem and give lower pressure drop than that of catalyst bed.
In this study, the electrolysis of water with hydrogen permeable Pd-Ag membrane electrode and hydrogenation of toluene from hydrogen permeated through the membrane in catalytic layer made of washcoated Pt/Al2O3 catalyst on stainless steel mesh. The potassium aqueous solution was supplied to reaction cell with HPLC pump at 0.8 MPa and toluene was supplied by bubbling with nitrogen at 0.1 MPa. The hydrogenation was conducted around 373 K. The stability of hydrogenation-electrolysis cell was stable for several hours. The effect of temperature and flow rate of toluene on hydrogenation rate and hydrogen utilization efficiency was evaluated, which revealed that the hydrogenation was enhanced in higher temperature region.
Syngas production had been great attention among the researcher especially from non-conventional feedstocks. Syngas is a basic feedstock for many commercial important chemicals and fuels and can be produced as a result by dry reforming of methane (DRM). However, conventional reactors involved high reaction temperature due to the high endothermicity of DRM. Membrane reactors are expected to decrease the reaction temperature by shifting the thermodynamic equilibrium of DRM. Several researchers have explored the performance of membrane reactors using metal and silica membranes in DRM, and confirmed hydrogen yield was improved. However, this reveals insufficient activity of conventional catalysts in low reaction temperature. Previously, we have developed Ru/Y2O3-ZrO2catalysts to beapplied to the low temperature DRM. In this study, we aimed to evaluate the effect of membrane performance on membrane reactor performance using our developed catalyst in DRM. In the case of the membrane reactor using a Pd membrane at 500 °C, CH4 and CO2 conversions were 32.5% and 25% respectively, which were a half-more higher than the conventional reactor. The hydrogen yield was found to be 33.9% that was three times higher than its conventional reactor, and interestingly,H2/CO ratio counted as 1.08 for the Pd membrane reactor and 0.70 for the conventional reactor. This indicates H2 separation gave a significant impact on not only the H2 yield but also H2/CO ratio. To compare with the Pd membrane reactor, Ni-doped silica membrane was also prepared by sol-gel method and its gas permeances were 0.53 × 10-6 mol m–2 s–1 Pa–1 for N2 and 3.20 × 10-6 mol m–2 s–1 Pa–1 for H2. The performance of the membrane reactor using the silica membranes will be presented as a comparison.
Key words : DRM, membrane reactor, syngas.
A great interest has concentrated to liquefied ammonia as a chemical hydrogen carrier in Japan because of its higher hydrogen content, i.e., 17.6wt%-H2 per unit weight of NH3. As far as ammonia is used as an energy carrier, its decomposition for obtaining H2 should be carried out at as low temperature as possible. However, the decomposition rate is known to remain very low at lower temperatures below 500 °C although the equilibrium conversion in the range 0.1-0.5 MPa at 300 °C reaches more than 80%. In practice, according to our data, the conversion remained around 10% at 300°C when 2wt% Ru/Al2O3 catalyst was used. Therefore, it is required to propose an innovative technology that can not only realize a higher conversion even at lower temperature below 400 °C but also obtain pure hydrogen.
As a way of improving such a situation, in this study, we have focussed on adoption of more active catalyst other than conventional Ru/Al2O3 and composite thin palladium membrane. A packed-bed palladium membrane reactor incorporating these catalyst and membrane is set up and then used for testing the ammonia decomposition at low temperatures.
First, it was found that Ru/CeO2 had a higher activity for ammonia decomposition when compared with Ru/Al2O3 catalyst at 350 °C. This is considered to be related to the basic property of CeO2 support itself, which may promote the recombinative desorption of N species. About 0.01 mm-thick palladium-silver alloy membrane supported on porous YSZ tube (11 mm in outer diameter) prepared with electroless-plating was used. The decomposition was carried out using the membrane reactors packed with either Ru/Al2O3 or Ru/CeO2. When comparing the performances between the conventional and membrane reactors at 300 °C, it becomes obvious that the conversion was increased by employing the Ru/CeO2 catalyst-packed palladium membrane reactor.
As an application of zeolite membranes to chemical reactions, various esterification reactions have already been examined mostly in liquid-phase perfectly mixed reactor to aim an equilibrium shift by selective water separation. Differently from such a batch operation, a vapor-phase flow one that makes efficient and continuous production possible, is considered to be advantageous. Dehydration reaction of alcohols, limited thermodynamically, is also industrially important and operated at higher temperatures, producing ethers together with water. This study focuses on vapor-phase dehydration reaction of ethanol to diethyl-ether, that is, 2EtOH → (Et)2O + H2O, which is equilibrium-limited. A membrane reactor incorporating chabazite (CHA) membrane and solid-acid catalyst is employed for removing water vapor selectively from the reacting mixture, thereby obtaining higher conversion.
CHA zeolite membrane was synthesized on a porous alumina tubular support (2.6mm in outer diameter and 105mm in length). A double tube membrane reactor, where around CHA zeolite membrane tube either granulated H-ZSM-5 or Amberlyst 15 (acidic ion exchange resin) as catalyst was packed, was built. Ethanol dehydration to diethyl ether was examined in the temperature range of 130 - 160 °C.
Prior to reaction, vapor permeation performance of the CHA membrane using an 80wt% ethyl alcohol-water mixture was evaluated at 150 °C. It was observed that the water content in the permeate was nearly 100wt% and the permeance was in the order of 10-7 mol/ (m2 s Pa). As the feed rate of ethanol decreases, i.e., the residence time increases, the reaction can proceed more. The yield of diethyl-ether in the membrane reactor is increased by 7% on the basis of the conventional reactor, where granular H-ZSM5 is used as catalyst. This is simply because more water vapour can be separated. More conversion is expected to be obtained when the more active Amberlyst instead of H-ZSM5 is used.
To decrease the pumping power for transporting thermal energy, a latent heat transportation system, which uses phase change material (PCM) slurries as a heat media has been attracted. Especially, the micro-encapsulation of PCMs is one of the promising techniques for preventing the pipe blockage due to the particle growth. The authors successfully fabricated silica hard-shell microcapsules containing PCMs. The microcapsules consisted of an inner hollow cavity and an outer SiO2 shell with porous structures. PCMs can be charged into the inner hollow cavity through the pores in the SiO2 shell. However, the charged PCMs are readily leaked out through the pore in the SiO2 shell during the utilization for the latent heat transportation system. To prevent leak of PCMs, the pores in the SiO2 shell are required to be covered with materials possessing micropores. In this study, the modification of ZIF-8 (Zeolitic Imidasolate Framework-8) film over an outer surface of spherical SiO2 was investigated.
Spherical SiO2 (CHROMATOREX) was obtained from Fuji Silysia Chemical Ltd. Japan. To facilitate the nucleation of ZIF-8 over the SiO2 surface, 3-(2-imidazolin-1-yl)propyltriethoxysilane (IPTES) was modified over the SiO2 in an aqueous solution of HCl at 333 K. The IPTES-modified sample was dispersed in a mixture of Zn(NO3)2 and 2-methylimidazole (Hmim) followed by still standing at room temperature for 24 h to form ZIF-8 film. The obtained sample was denoted as ZIF-8/SiO2.
From the result of XRD measurement, ZIF-8 structure was formed at the Hmim/Zn molar ratio of 30 during the preparation process of ZIF-8/SiO2. The roughness surface was observed over the ZIF-8/SiO2 by SEM. From the mapping of Zn obtained by SEM-EDX analysis, Zn species existed over the outer surface of SiO2. These results suggest that the ZIF-8 film can be formed over the outer surface of SiO2.
Acknowledgements: This study was supported by JST-Mirai-project: #JPMJMI17EK.

In order to obtain the value-added cheicals ethyl cynnamate, this produce should be synthesized with high stereoselectivity. The Horner-Wadsworth-Emmons (HWE) reaction is a promising reaction to obtain the ethyl cynnamate. The high selectivity of HWE reaction required the sever low temperature or basic conditions. To overcome these severe reaction condition, the effect of liposome membranes as a reaction field was examined because liposome that is a closed lipid bilayer exhibits the enhanced-effect of reactivity for some reactions [1].
In this study, ethyl diethylphosphonoacetate and ethyl diphenylphosphonoacetate were used as E-prefer and Z-prefer substrates for of HWE reaction, respectively. The HWE reaction was, at 25 oC for 8 hours, performed to mix the substrate with benzaldehyde in the presece or absence of K2CO3, sodium iodide, 1,8-diazabicyclo[5,4,0] undec-7-ene (DBU), and liposomes. The obtained products were analyzed by a 1H-NMR for a quantification of E- and Z-isomers.
The effect of liposomes on the stereoselectivity of two substrates was tested. Use of the liposomes existing in the gel phase resulted in the enhanced activity of the substrates and furnished the products with same E/Z stereoselectivity as in the liposome-free system. The membrane environment in the gel phase most likely assisted the formation of adducts that induced selective generation of the E-isomer. It is noted that the energy barrier for the Z-isomer could be reduced by the addition of liposomes. Therefore, the observed effects of liposomes were likely to result from the use of their voids for the stable formation of intermediate adducts and their activity similar to the basicity of DBU. Figure shows the case of ethyl diethylphosphonoacetate. The possible role of liposomes is to assist the proton removal from the reactant, rather than providing the basic interfacial environment.
[1] T. Shimanouchi et al., J. Biosci. Bioeng., in press.
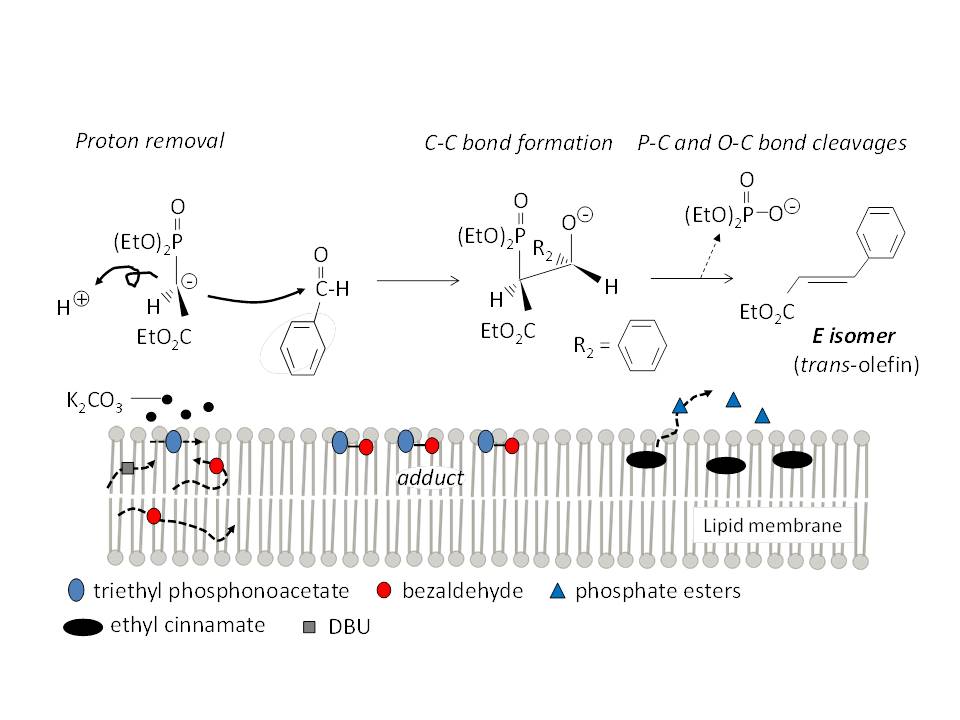
The transparent conductive film is a thin film having conductivity with an average transmittance of 80% or more in the visible light region and a resistivity of 10 -3 Ωcm or less, is used for flat panel displays and the like. At present, indium tin oxide is mainly used as a transparent conductive film, but since the price is so high due to concern about depletion of indium, development of alternative materials is required. Zinc oxide thin films are known as transparent conductive films, are reported to be further lowered in specific resistance by adding aluminum to form aluminum-doped zinc oxide (AZO), and thus are expected as alternative materials. Thermal CVD is a surface coating method in which substances in the gas phase are deposited on the substrate in solid form using chemical reactions on the surface of the substrate, and heat is applied to promote the chemical reaction to raise the temperature of the substrate. And, it is mainly used for oxide film formation. Further, by reducing the pressure in the apparatus, the mean free path of the reaction gas becomes longer, and the film thickness uniformity is improved. Therefore, an AZO thin film was prepared by a low pressure thermal CVD method, the average transmittance in the visible light region and the resistivity were measured, and the relationship with the film forming conditions was studied.
Silicon nano particle is a promising material as an electrode of Li ion rechargeable battery. Productivity of the silicon nano particle is low, so improvement of the productivity is required. We have investigated combination of plasma enhanced Chemical vapor deposition (PE-CVD) and high speed jet as a gas supply method in order to obtain high deposition rate of silicon material. In this study aggregated silicon nano particles were rapidly deposited on cupper substrates using Plasma Enhanced CVD in SiH4/H2 system to improve productivity of silicon nano particles.
In our experiments input power and frequency of the power supply was 50 W and100 MHz respectively, and pressure of an experimental vacuum chamber was 800 Pa. The deposited particles was evaluated by SEM image. Deposited mass was measured by an electronic balance. Crystallinity was analyzed by Raman spectroscopy. Deposition thickness and surface roughness was measured by a laser 3-dimensional measuring instrument.
Figure shows SEM image of deposited silicon particles. Diameter of the silicon particles are several tens of nm. Thickness of silicon materials by 1 min deposition is several tens of micrometer. Raman analysis show the deposited particles are mixture of amorphous and nanocrystalline silicon.
Relation between deposited mass of silicon and both distance of two electrodes and mass flow rate of the source gas was investigated in order to obtain better productivity of the nano particles.
Velocity of the source gas introduced into a vacuum chamber was several hundreds of m/s in our experimental setup. The experimental result shows that the nano particles were generated during convection in the vacuum chamber after and transported to substrate by the jet of source gas.

Atmospheric-pressure plasma jets (APPJ) have recently been a topic of great interest. Especially, dielectric barrier discharge (DBD) jets, which can be driven at room temperature come to play an increasing role on process of organic materials which are sensitive to heat and biomedical applications. Cancer treatment, sterilization and dental treatment are example.
For less invasive APPJ optimization, we tried to develop a new type of jet which can process objects at low temperature and high speeds. This APPJ can generate stable, arc-free discharge with only air plasma gas. High voltage electrode was Ti wire and coaxially inserted into nozzle. Material of the nozzle was quartz tube. Inner diameter of the quartz tube was 4mm, and outer diameter was 6mm. Ground electrode was Ta foil with irradiation hole and placed at the tip of the nozzle. Size of the irradiation hole was 1mm. Voltage applied was 4kVp-p, and the frequency was 25kHz. Feed gas was dry air, and the flow rate was 5, 7.5, 10slm. The polymer film based on poly-methyl methacrylate (PMMA) was used to etch, placed downstream from the jet nozzle. Irradiation time was 60s. At each flow rates, etching was performed at a distance such that the temperature of the irradiated object was about 40°C(Jet to object distance was; 7mm at 5slm, 4mm at 7.5slm, 3mm at 10slm). As shown in the figure, the results were 0.05μg/s/W at 5slm, 0.11μg/s/W at 7.5slm and 0.13μg/s/W at 10slm. From this result, temperature may not significantly affect etching. The effects of thickness and hole size of the ground electrode were also evaluated.
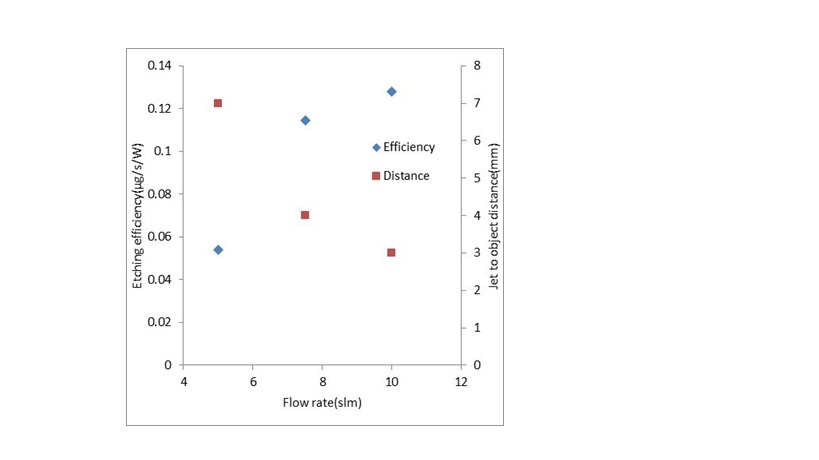
Carbon nanotubes (CNTs) have various excellent properties and potential applications. When synthesizing CNTs, metals such as Fe are normally used as catalyst, which need be removed for many applications. Wet process using acid solution is widely used as removal, which causes difficulty in redispersion of CNTs due to strong agglomeration upon drying. In addition, oxidation is also often combined to the acid etching to remove the carbon shell covering the catalyst particles, which causes damage to CNTs. To solve these problems, dry process has been developed in which halogen gas is flowed through CNT powder at high temperature and remove catalyst metals as metal chlorides. However, halogen gas is highly dangerous and require special facilities, thus challenges remain in industrialization. Here, we propose a highly safe process using FeCl3, which is solid and has negligibly small vapor pressure at normal temperature and pressure. CNT powder by floating catalyst chemical vapor deposition (CVD) and FeCl3 power were set apart in a quartz glass tube, the tube was filled with Ar and heated using a furnace. FeCl3 evaporates and diffuses through the CNT powder, removing Fe catalyst by the reaction of Fe (s) + 2FeCl3 (g) → 3FeCl2 (g). Over 80 % of Fe was removed from CNT by scanning electron microscope-energy dispersive X-ray spectroscopy measurement (Fig. 1). Raman spectra showed the retained intensity ratio of G-band to D-band peaks, showing CNT was not damaged (Fig. 1). From TEM images, CNTs had metal particles covered with carbon shells before purification (Fig. 1) while had empty carbon shells after purification (Fig. 1). Furthermore, this method proved effective for various CNTs, removing Fe catalyst from multi-wall CNTs synthesized by supported catalyst CVD and removing Ni-Y catalyst from single-wall CNTs synthesized by arc discharge method.
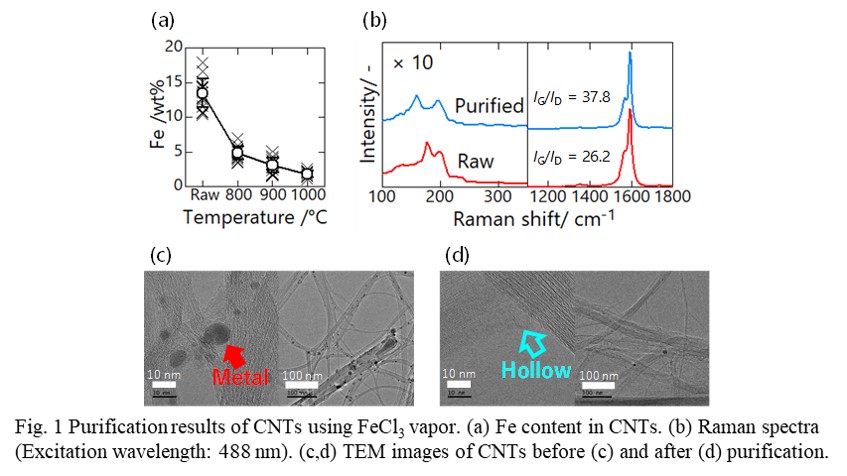
Carbon nanotubes (CNTs) have been extensively researched owing to their unique one-dimensional nanostructure and properties. Mass production has been established for multi-wall CNTs (MWCNTs) instead of single-wall CNTs (SWCNTs). Floating catalyst chemical vapor deposition (FCCVD) yields high-quality SWCNTs [1,2] but the productivity is small because individual SWCNTs are much lighter (~1/10,000 of 150-nm-diameter MWCNTs of the same length). Generally, catalyst precursors are fed to externally heated reactors, and heated in >1 s, and catalyst particles gradually nucleate at low density. We have previously proposed and developed the flame-assisted CVD (FACVD) method [3], in which a premixed flame is used to heat and mix the gas quickly in ~1 ms. The FACVD method yielded 1 nm-diameter, high-quality SWCNTs. However, the flame requires oxygen at high concentration, causing narrow growth window and small productivity (~0.3 mg-CNT/min).
In this work, we report the FCCVD method with a micro-nozzle for preheating catalyst precursors. We used ferrocene and sulfur vapors as catalyst precursors, preheated and mixed them with CH4/H2/Ar, and synthesized SWCNTs at 1200 C. Product is yielded as black smog continuously from the reactor (Fig. 1a). The product was clean ropes of SWCNTs with few particles (SEM, Fig. 1b) with a Raman spectrum characteristic for high-quality SWCNTs (sharp G-band peak, high G-band to D-band peak intensity ratio of 102, and strong RBM peaks, Fig. 1c). The thermogravimetry analysis in air showed the ash content of 6.66 wt%, corresponding to carbon purity of >95 wt%. A productivity of 8.0 mg-CNT/min and carbon yield of 4.2% were achieved using a lab-scale reactor (inner diameter of 40 mm).
[1] A.G. Nasibulin, et al., Chem. Phys. Lett. 402, 227 (2005).
[2] T. Saito, et al., J. Phys. Chem. B 110, 5849 (2006).
[3] S. Okada, et al., Carbon 138, 1 (2018).
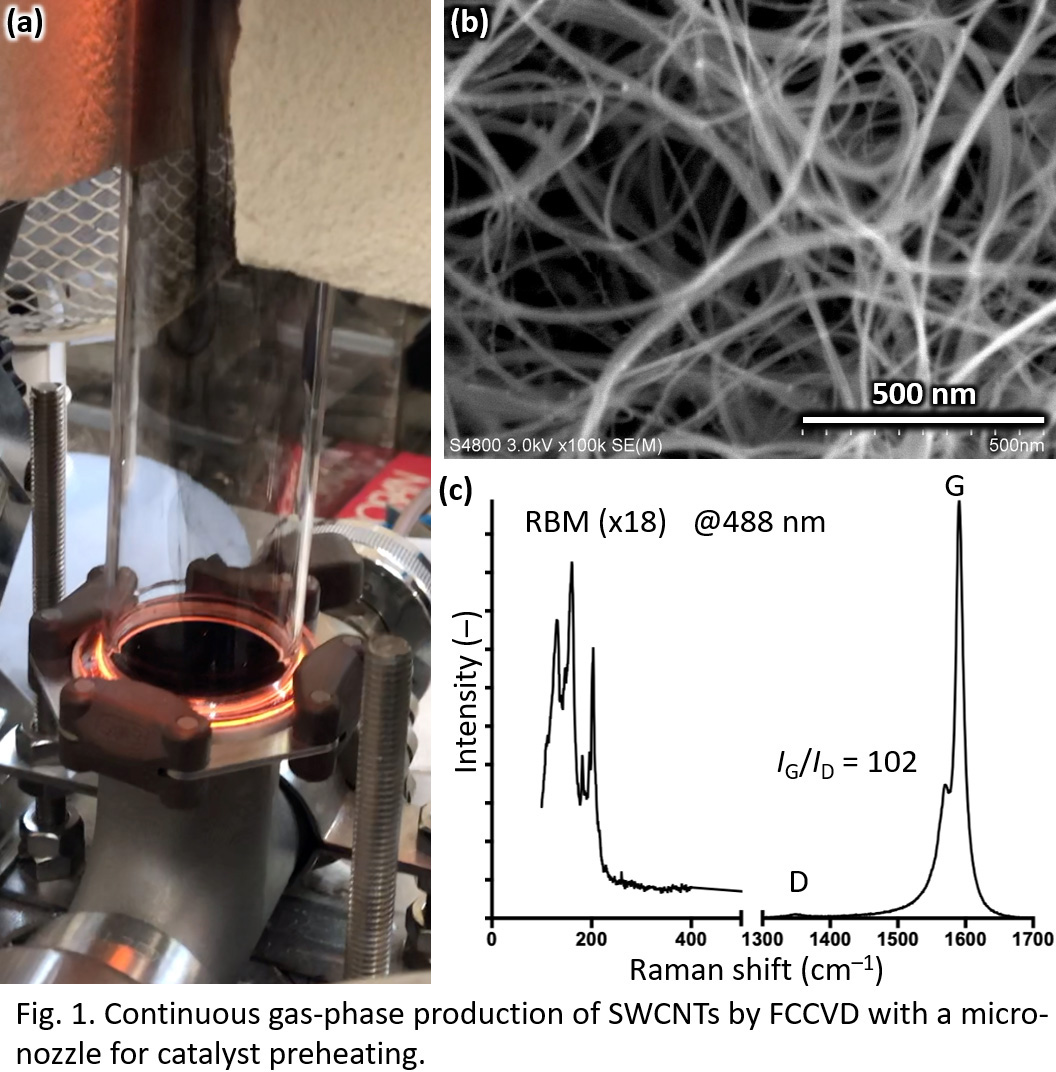
Carbon Nanotubes (CNTs) are attractive with high hopes in many different fields for their excellent properties. However, the practical applications of CNTs has been a challenge due to their high price and/or insufficient quality. Floating catalyst chemical vapor deposition (CVD) enables the synthesis of high quality CNTs, but it has had an issue with its low catalyst usage. To combat this situation, many groups have conducted research to improve the productivity of this process, and as a result it has been reported that the implementation of sulfur can increase the overall yield of floating catalyst CVD [1]. Many groups have proposed positive roles of sulfur; Fe-S to be the active site for catalytic reaction or reduced surface energy of Fe-S for nucleation of small Fe particles. However, these models are not supported by clear evidences and are not applicable to supported catalysts.
Here, we propose an opposite model; sulfur deactivates the Fe catalyst, small amount of sulfur forms an inactive region on a Fe particle, and the inactive region works as an entrance for the carbon source without being covered with graphitic carbon. The analysis of this is difficult for floating catalysts, therefore for this research, supported catalysts were used by implementing graphite sheets as substrates to replicate the floating catalysts of spherical structure. Scanning electron microscope (SEM) shows that Fe on graphite grew no CNT without sulfur but many CNTs with sulfur (Fig. 1a). To confirm the importance of having inactive region within the catalyst particle for CNT synthesis, thin Al2O3 layer were deposited on top of the Fe catalysts. The thin Al2O3 layer significantly enhanced the CNT growth (Fig. 1b) in a similar manner as sulfur. Our new concept enhancing catalyst activity by making inactive region on catalyst particles enables new catalyst design.
[1] H.M. Cheng, et al., Appl. Phys. Lett. 72, 3282 (1998).

Chemical vapor deposition (CVD) has been widely used for fabrication of functional films. Solving mass transfer of relevant chemical species and their chemical reactions to make deposition during CVD is essential to predict the film thickness profile thereby to design an optimal reactor without resorting to empirical approach. As the figure shows, sometimes the substrates for CVD have microstructures on the surface, but solving the equations in a single geometry that includes both reactor and microstructures is unfeasible due to scales difference. Accordingly, microstructures have been separated from the reactor, where equations were solved individually and iterated until boundary condition of their interface becomes identical. However, it always imposes repeated calculation, which cannot be implemented in the commercial software, thus troublesome. Here, we propose a concise low-cost multiscale simulation method by using COMSOL, a software basing on finite element method. Microstructures and reactor were separated likewise iteration method, while the equations were concurrently solved by coupling the geometries with boundary conditions expressed by equations. The boundary conditions are concentration and flux of each species. The concentration was set to be continuous. The flux was defined as integral of deposition rate in the microstructures because total consumption rate of the precursor in the microstructures equals to the precursor flux across the interface. Hence, automatic low-cost multiscale simulation was enabled. Then, we imparted time-dependent simulation capability to cope with shape change of the microstructures by the deposited film. Growth rate profile in the microstructures obtained above was used to update the shape, which will be used for simulation in next time step, by level set method. By repeating this at each time step, time-dependent multi-scale simulation was enabled. We applied this methodology on W deposition to get profile evolution successfully and the result showed the importance of feedback by microscale topography.