For the practical use of Lithium-air batteries (LABs), their cycle performance must be improved. In this system, oxygen supplied from the atmosphere reacts with Li+ and Li2O2 is deposited in the cathode during discharging. To obtain a large capacity and a high rate performance, the cathodes need to have a high surface area and a high electrical conductivity. Therefore, porous carbons are generally used for the cathodes.
To improve cycle performance, side reactions, which involve the decomposition of the electrolyte and result in irreversible capacity, need to be suppressed. It has been reported that oxygen functional groups existing on carbon materials, promote such side reactions. On the other hand, it has been also reported that amorphous Li2O2 tend to be formed on carbon materials having oxygen functional groups more than pristine carbon materials. Such amorphous Li2O2 easily decomposes during subsequent charging, and the charge over potential is relatively low. These facts indicate that there is a suitable amount of oxygen functional groups to increase cycle performance, which has not been clarified.
In this study, the quantitative effect of oxygen functional groups on cycle performance was investigated using carbon gels (CGs) as a model carbon material. CGs have a large mesopore volume which can contribute to the capacity of LABs, and their surface properties can also be tuned. While CGs originally have many oxygen functional groups, they were reduced by heat treatment at various temperatures. Then, the oxygen functional groups were quantitatively evaluated by temperature programed desorption analysis (TPD) performed under vacuum. The results obtained using CGs heated at 2200 °C showed that most of the functional groups were removed at this temperature and the resulting carbon can maintain a discharge capacity of 500 mAh g-1 for 21 cycles, while CGs heated at 1000 °C could maintain this capacity only for 8 cycles.
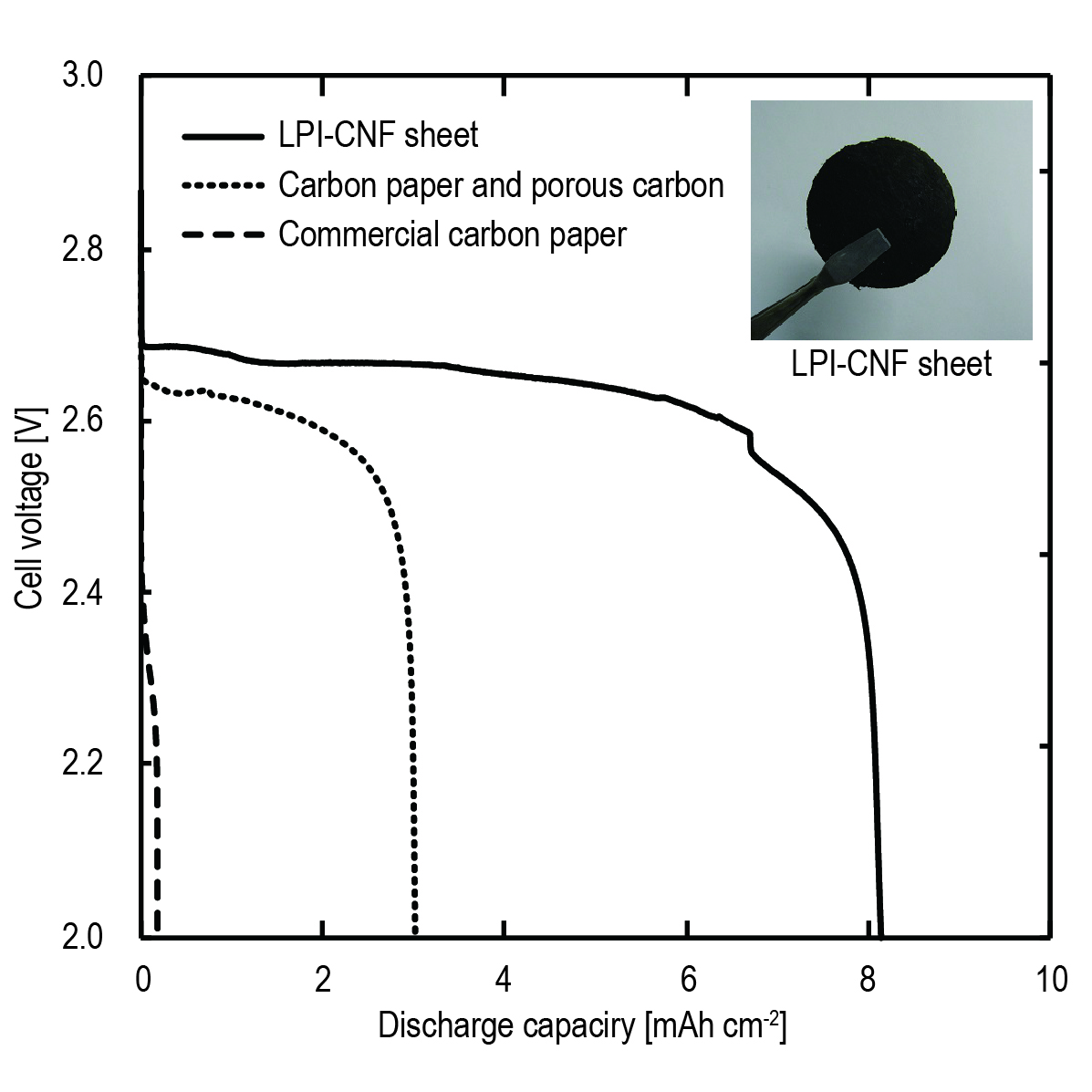
Lithium-air batteries are expected to become a secondary battery of the next generation owing to their high theoretical energy density. Carbon paper combined with a porous carbon are generally used as the cathode in this system because the surface area of a typical carbon paper is too low to be used independently. In contrast, carbon nanofibers (CNFs) with a fiber diameter of several tens of nanometers have a sufficient surface area for this purpose. In this study, sheet electrodes were developed using CNFs produced through the liquid pulse injection (LPI) technique. The CNFs can be efficiently synthesized and easily casted into a sheet electrode only by vacuum filtration. Cathode performance of the sheet electrodes for lithium-air batteries was evaluated and optimal conditions to obtain higher capacities were explored.
LPI-CNFs with a diameter around 20 nm were dispersed into ethylene glycol. The resulting dispersion was filtered under vacuum to form a sheet. Coin cells of 2032-type were assembled using the sheet and a solution of CF3SO4Li in tetraglyme (1:4, molar ratio) as the cathode and electrolyte, respectively, and the cells were galvanostatically discharged and charged in an oxygen atmosphere.
The LPI-CNF sheets had a mechanical strength similar to that of a commercial carbon paper and much higher surface area than that of them, suggesting that the LPI-CNF sheets can be used independently as cathodes.
From results of discharge/charge measurements, the LPI-CNF sheets showed about a 3 times larger discharge capacity than that of an electrode prepared from a carbon paper and porous carbon.
It has been reported that usable depth of electrodes is limited because of the influence of oxygen diffusion. Thus, the structure of the LPI-CNF cathodes such as porosity and thickness were optimized to increase discharge capacity.
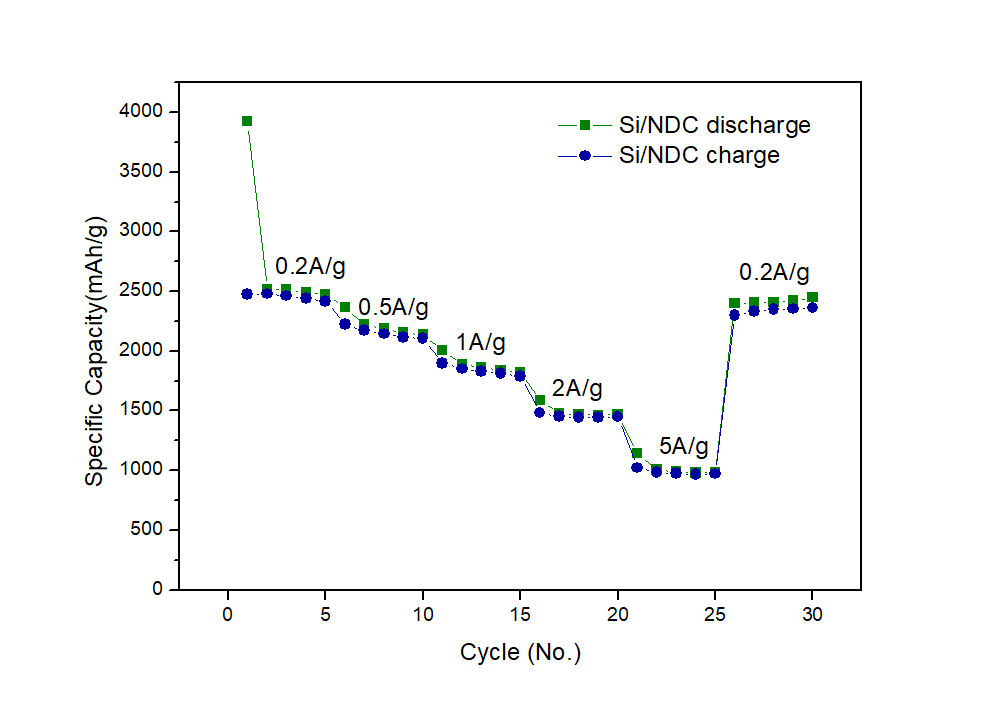
Lithium ion storage devices are indispensable for modern society because of their high energy densities, low self-discharge, no memory effects, and applications in many high-tech industries such as electronic devices, military, and battery powered vehicles. However, most commercial lithium ion storage devices use carbon electrodes, which have low theoretical capacity (≈ 372 mAh g-1) and are not expected to meet the future needs of the energy storage industry. Consequently, silicon, possessing high theoretical capacity (≈ 4200 mAh g-1), being abundant in nature, and exhibiting low discharge potentials, has become a promising anode material. Despite these advantages, the use of Si anode is hindered by its low electrical conductivity and large volume changes during the lithiation/de-lithiation cycles leading to subsequent pulverization. Herein we designed a porous composite nanostructure of silicon with N-doped carbon (NDC) from mesoporous silica sphere and dopamine, which can offer space to accommodate the volume changes and improve the electrical conductivity of the electrode for performance enhancements. Indeed, the as-synthesized Si/NDC composite delivered a high capacity of 2,518 mAh g-1 at a current density of 200 mA g-1 and maintained a high capacity of 980 mAh g-1 at a high current density of 5,000 mA g-1.
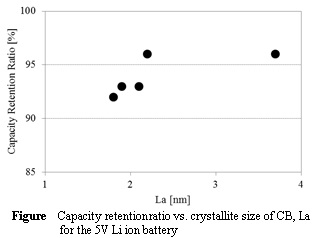
Side reactions on high-voltage cathode for lithium ion batteries would be influenced by physicochemical properties of carbon black (CB) conductor used in it. We have prepared various CB conductors with different surface characteristics and crystallinity, and have applied them to a 5V cathode, LiNi0.5Mn1.5O4 (LNMO) as conductors. By electrochemical evaluation some samples were found to be suitable for the 5V cathode. The reaction mechanism would be reported.
At the present time, ternary cathode materials, NCM, have attracted many researchers to study due to their excellent electrochemical properties, such as high specific capacity and high charge-discharge voltage. By tuning the portions of nickel, cobalt and manganese, NCM can obtain different electrode characteristic. However, their poor stability when worked at high operating voltage have limited their applications in industry. In this study, polyimide (PI) was covered on the surface of NCM523 cathode material by a simple liquid phase modification. At first, 2,2-Bis(3-amino-4- hydroxyphenyl)hexafluoropropane (BisAPAF) and 2,3,3,4-oxydiphthalic anhydride (ODPA) were mixed in the solvent, N-Methyl-2-Pyrrolidone (NMP), at room temperature to synthesize the polyamic acid (PAA). Then, a desired amount of NCM523 powder were added and dispersed in PAA solution by stirring for 4h to obtain NCM523/PAA material. Finally, the NCM523/PAA material was converted into NCM523/PI by multistep heating process. XRD analysis showed that the addition of PI did not affect the original layered structure of NCM523 cathode material. The energy dispersive spectroscopy (EDS) analysis showed that polyimide could be evenly distributed on the surface of NCM523 particles. TGA analysis also confirmed a slight weight percentage of PI was added to the NCM523 material. The effect of PI addition on the electrochemical properties of NCM523 cathode material is still under investigation, and it is hoped that the present polyimide (PI) coating can effectively prevent the undesired interfacial reaction between the electrode material and the liquid electrolyte.
All-solid state batteries (ASSBs) are expected as next-generation battery for electric vehicles (EVs), hybrid electric vehicles (HEVs) because it is safer than usual liquid-type Lithium-ion battery. Recently, the innovative solid electrolyte have been developed, and it is comparable to organic liquid electrolyte in ionic conductivity. However, it is desired to attain more power density and more energy density. In order to improve the performance of ASSBs, it is required to optimize an electrode structure as well as material characteristics such as active materials and electrolytes. Therefore, monitoring the state in electrode layer during charge and discharge using the numerical computation is a critical measure to understand the phenomena in a cell. In the relatively micro-scale system such as the electrode layer, even a slight difference in structure remarkably affects the battery performance. However, usual simulations employ the empirical overall characteristics such as the reactive interface area and the tortuosity factor, so the phenomena resulting from minute structure of electrode layer which notably affect the cell performance might be overlooked. Hence, in this study, the numerical computation technique that can reflect the micro-characteristics of electrode structure directly by building a multi-element network model based on porous structures was developed. And the effect of heterogeneous electrode structure on cell performance was considered.
In this study, 0.5 M nickel hydroxide, cobalt nitrate, and manganese nitrate were used as the metal sources with molar ratio of Ni:Co:Mn=1:1:1 and sodium hydroxide as the precipitant to prepare the NCM(OH)2 precursor by co-precipitation for application in the synthesis of NCM ternary cathode materials. The volume flow ratio of metal nitrate and NaOH was 1:2 and the pH value of the precipitation reaction was controlled at 10-12. After the reactants were mixed, the mixture was stirred at 50 °C for 12 h under nitrogen atmosphere. It was found that the color of the precipitate gradually changed from grey to brown in the experiment. It was speculated that the color change was resulted from the formation of manganese oxide-hydroxide. It was known that the manganese hydroxide could be reacted easily with trace oxygen even in a nitrogen atmosphere. SEM images showed that the precursor powder obtained by the preparation was irregular granular and had the obvious aggregation phenomenon. Besides, the XRD analysis showed that the precursor powder of the ternary positive electrode material was successfully prepared in this study and the precursor was composed of nickel cobalt manganese-hydroxide NCM(OH)2 and nickel cobalt manganese-oxygen-hydroxide NCMO(OH)2.
Vanadium redox flow batteries (VRFB), which use vanadium species as the active materials, have advantages such as the flexible design between power and capacity. Thus, VRFB is expected to store renewable energy. However, the current density of a single cell is small due to the higher internal resistance such as reaction resistances. Therefore, researches have been conducted to reduce the reaction resistance by addition of surface oxygen functional groups by heat treatment in air or liquid phase oxidation to the carbon electrode material. However, the conventional treatment methods have issues such as longer treatment time and necessity of waste liquid treatment. In consequence, in this research, we focused on electron beam irradiation as the effective oxidation method. It is considered that the surface oxygen groups can be added by the reaction between surface carbon and radicals and/or ozone from the water or the oxygen formed by the irradiation. This processing method can be performed in a short time of 60 minutes or less at room temperature.
In this study, Electron beam irradiation was performed on the carbon cloth, then, characterization and electrochemical measurement were performed. Two-sheet of Carbon cloth (CC) was used as the electrode material. The electron beam with acceleration voltage of 2.0 MV was irradiated to the CC under air atmosphere at Takasaki Advanced Radiation Research Institute. To evaluate the cell performance, current-voltage curves were obtained using a single cell with interdigitated flow channel. The current density of the irradiated electrode under air was improved compared with the non-irradiated electrode. X-ray photoelectron spectroscopy (XPS) measurements were performed to analyze surface oxygen groups. As a result, the surface oxygen, i.e., C-O, COO, etc., of electrode material increased by electron beam irradiation within 60 minutes.
Supercapacitors are one of the most promising energy storage devices because of their long cycle life, high power densities, and high discharge efficiencies. The power and energy densities of supercapacitors depend critically on the electrode materials. In recent years, metal-organic frameworks (MOF) have attracted much research attention as electrode materials for supercapacitors, because of their unique properties such as high porosities, large surface areas, and tunable morphologies, which are beneficial to the capacitive performances of supercapacitors. In this work, we synthesized Mn-based and Mn-Fe based MOFs via a simple solvothermal method, using 1,4-benzenedicarboxylate as the organic linker. The incorporation of Fe into the Mn-based MOF significantly enhanced the capacitive performances. The specific capacitances of Mn-based MOF and Mn-Fe based MOF were 21 and 75 F/g, respectively in 2M KOH at a scan rate of 5 mV/s. The enhancement is attributable to the synergistic effect between Mn and Fe.
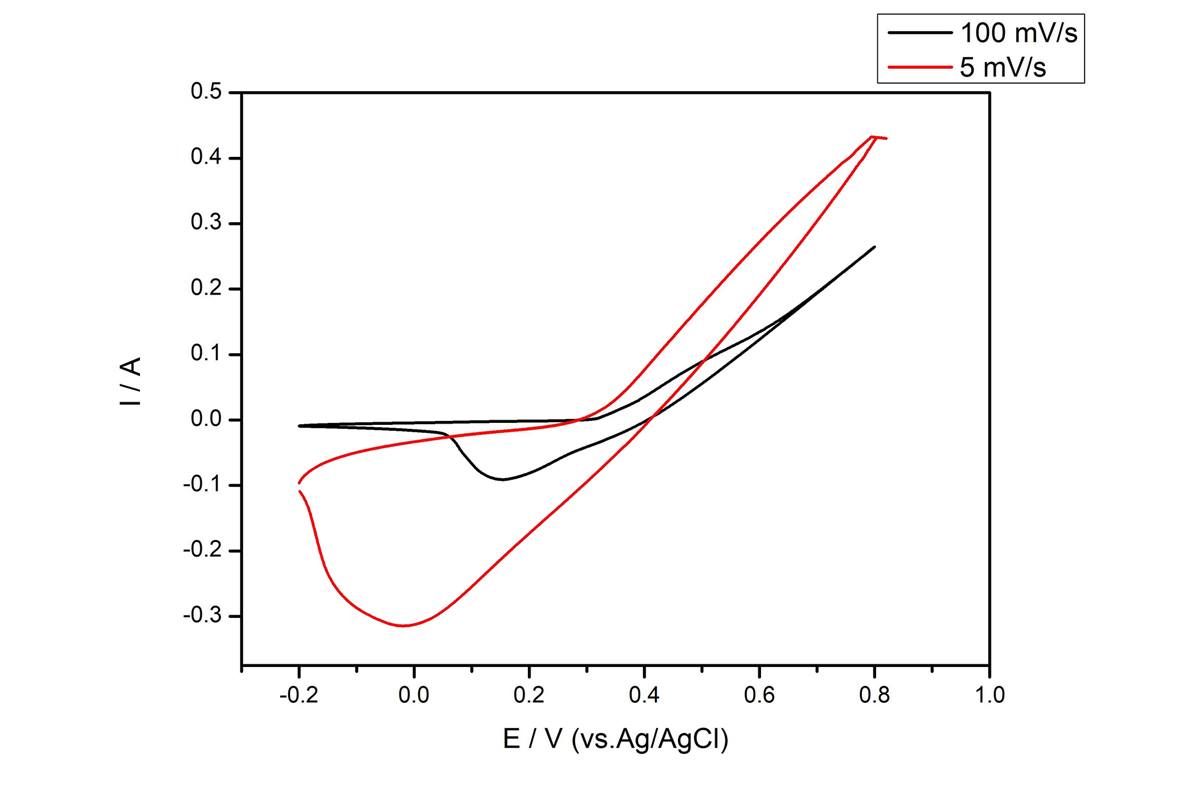
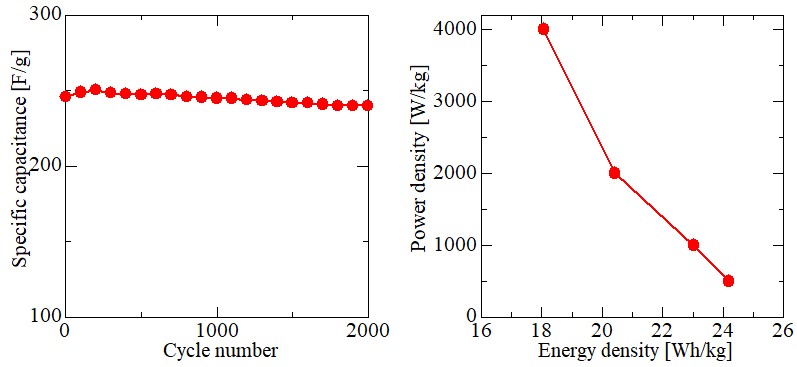
Recently, Microwave is an efficient, green and selective heating method, it has been employed to reduce graphene oxide (GO) in solvent or under dry condition. Firstly, we used modifification of Hummers method to fabricate graphene oxide (GO), then graphene oxide (GO)/Titanium oxide (TiO2) nanocomposites were synthesized with different wt/wt % of GO/TiO2 (1:1; 1:2; 1:3; 1:4 and 1:5), finally the nanocomposites were reduced by microwave method. To assess the properties of materials for use in supercapacitors, cyclic voltammetry and galvanostatic charging–discharging measurements were performed. And the results show that the microwave method synthesis of reduced graphene oxide (GO)/Titanium oxide composites with wt/wt % of GO/TiO2 (1:4) has the highest specific capacitance of 454 F/g at a scan rate of 10 mV/s. What's more, as shown in Figure 1, it has a high energy density of E = 24.2 Wh/kg and high power density of P = 500.1 kW/kg. As a result, graphene-based material obtained by MW-assisted reduction has a porous and loose structure and relatively high special surface area, making it perfectly suitable for supercapacitor devices.
For widespread use of renewable energy, effective utilization of excess energy from the renewable energy is a key issue. Electrolysis of hydrogen carrier, especially ammonia, is a possible solution because ammonia possesses some advantages including stable chemical property, high hydrogen density, easier storage and non-explosion. Unfortunately, during electrochemical reaction of ammonia synthesis with metal catalysts, hydrogen evolution reaction also occurs, and thus the faraday efficiency of ammonia electrosynthesis becomes low. In this study, the effect of addition of tungsten into iron on improving electrochemical reaction of ammonia synthesis and suppressing hydrogen evolution reaction were investigated using 90%N2-10%H2, W-Fe-BaCe0.9Y0.1O3 (BCY)|BCY|Pt, 3%H2O-77%Ar-20%H2. W-Fe-BCY catalyst cathode was fabricated by the incipient wetness co-impregnation method. After addition of W into Fe catalyst, the current density decreased because of larger energy of tungsten hydride formation which suppresses hydrogen evolution reaction. Impedance spectroscopic analysis also suggested the same result of suppression of hydrogen evolution reaction. Furthermore, the ammonia formation rate became higher at lower operating temperatures than those of pure Fe catalyst. This was probably caused by lower coverage of hydrogen adatoms on W-Fe catalyst surface and retaining active sites for N2 dissociation. In addition, with an increase in ammonia formation rate and a decrease in current density, the faraday efficiency also increased by around two times with the addition of W into Fe catalyst.
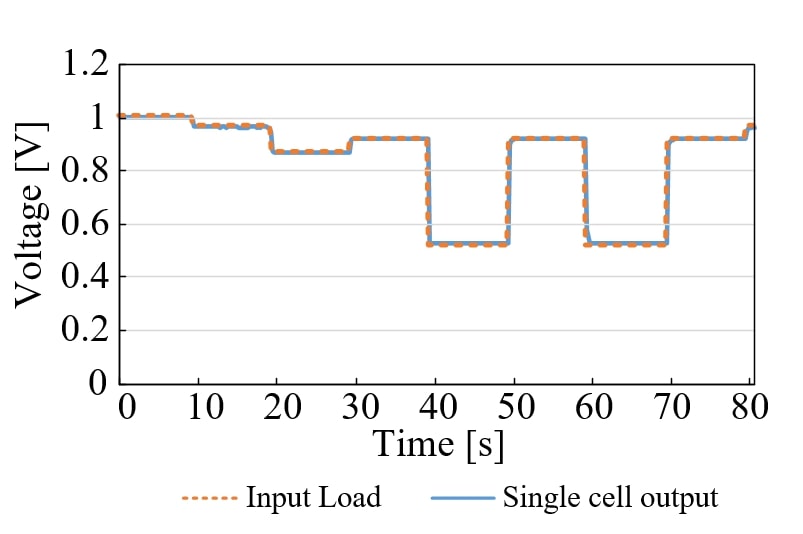
Recently, Global warming has become a serious problem. Therefore, Japan is aiming at a hydrogen society and conducting demonstration experiments of hydrogen supply chains in various places. Fuel cells are the most popular source of hydrogen energy. However, the load followability of fuel cells has not been studied. In this study, we will construct a power generator using the SOFC single cell which is the highest temperature operation and the highest efficiency among fuel cells, and investigate the load following ability. Currently, it is desirable to make SOFC operable at low temperatures. Therefore, in this experiment, the operating temperature is 700 degrees, and the flow rate of each gas is 200 mL/min. First, the I-V characteristic of a single cell is measured in order to investigate whether it is a suitable experimental device. From the experiment, the power per unit area at peak was 0.1 W/cm2. Since this power value is equal to the nominal value, it can be said that an appropriate experimental device has been produced. Next, based on this value, a variable load is created using the programmable load function of the DC electronic load. The loading rate is repeated at 20% and 100% every 10 seconds from 40 seconds to 80 seconds. The single cell output when the created variable load is input to the single cell is shown in the figure. From the figure, it can be seen that in any case, tracking follows less than 0.6 seconds. From the experimental results described above, it is found that the SOFC single cell has high load followability. In the future, CO2 is expected to be reduced by replacing the thermal power generator with a SOFC generator for the peak load power supply.
Anode materials based on transition metal phosphides were investigated for intermediate temperature fuel cells (ITFCs). Metal phosphides are world-widely investigated as highly-active and selective catalysts for hydrodesulfurization, hydrodenitrogenation, and hydrodeoxygenation. These reactions require catalytic ability for dissociation of H-H bond as well as C-S, C-N, or C-O bond. In addition to the catalytic activity, metal phosphides are known to be thermally and chemically stable and to possess electric conductivity comparable to metals and hardness similar to ceramics. Because of these features, metal phosphides are expected to function as electrodes in electrochemical cells. We have reported that molybdenum phosphide and tungsten phosphide as the anode of ITFCs demonstrated high power generation characteristics per electrochemical surface area. Accordingly, this study aimed to increase electrochemical surface area by supporting metal phosphides on carbon materials. Also, to increase the porosity of the anodes for ITFCs, carbon black (CB) and carbon nanofiber (CNF) were mixed with commercial platinum carbon (Pt/C) catalysts. Power generation and cyclic voltammetry were conducted to evaluate the catalysts' electrochemical activities. From the results of power generation and CV, molybdenum phosphide loaded on CB shows higher power generation characteristics per electrochemical surface area than Pt/C. It is suggested that phosphides loaded on carbon were well dispersed and many active sites were produced. Power generation measurements revealed that Pt/C and CB with the weight ratio 1: 1 and Pt/C and CNF with the weight ratio 1: 1 exhibited good cell performances. It indicates that increased porosity in the electrode gave rise to large triple-phase boundary.
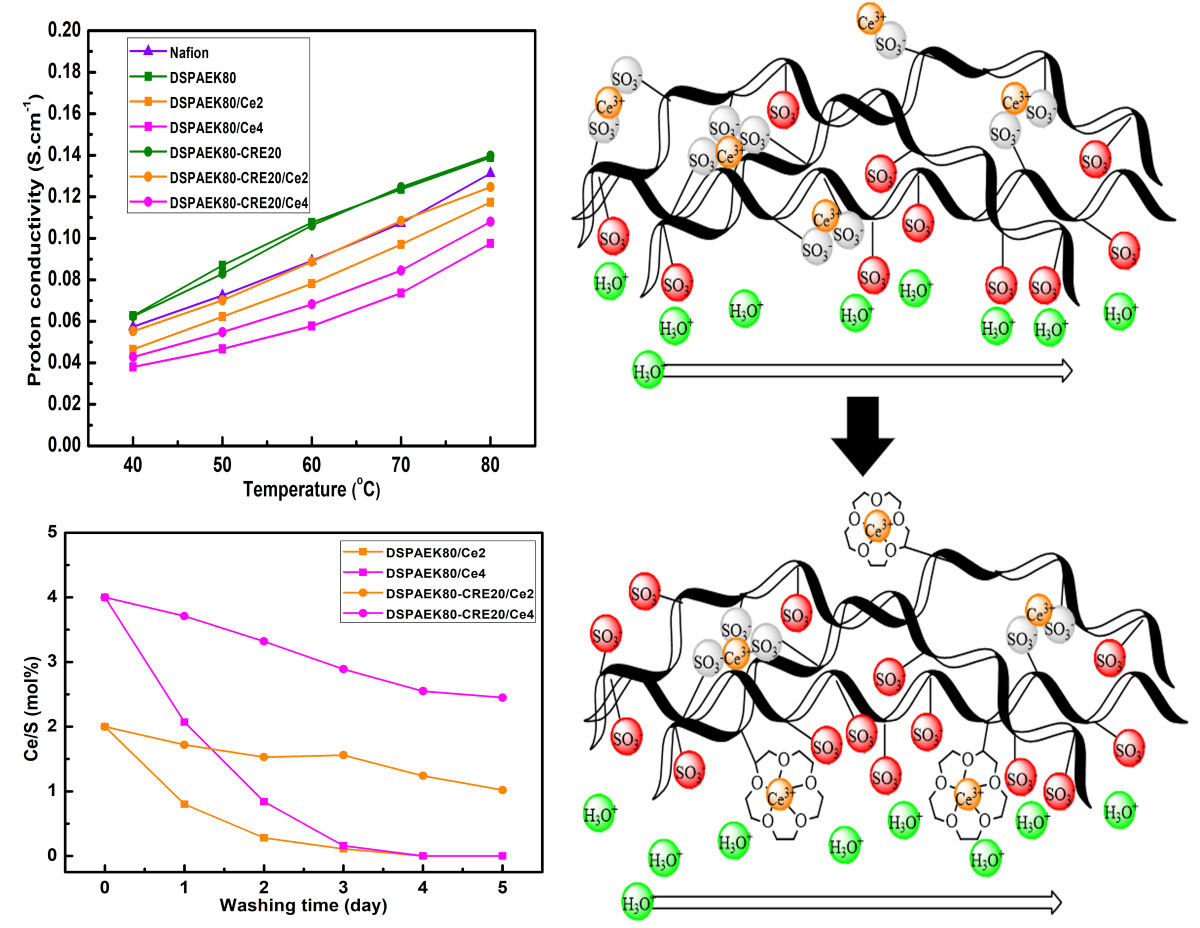
In the proton exchange membrane fuel cell, the durability is recently the critical issue in its operation. As the Ce+3 cerium ion (Ce) is a potential scavenger for ·OH and ·OOH radicals, the main causes of the chemical degradation of the membrane, aminomethyl-15-crown-5-ether (CRE) and dual sulfonated 3,3-diphenylpropylamine (DSDPA) were grafted onto the poly(arylene ether ketone) (PAEK) to maintain high proton conductivity and chemical stability by alleviating the migration of Ce from the membrane. The chemical and physical structures of the synthesized CRE grafted dual sulfonated PAEK membrane (DSPAEK-CRE) along with the coordination complex of CRE and Ce ions were investigated using FT-IR, 1H-NMR and SAXs, and XPS spectroscopy. The Fenton's test showed huge improvement of anti-oxidation stability by the presence of CRE. While the CRE/Ce coordinated membrane showed higher proton conductivity and better chemical stability than the Ce+3 dispersed one, other properties such as water uptake, swelling ratio, and mechanical strength were not significantly affected by CRE.
Recently, solid-state alkaline fuel cells (SAFCs) with anion-exchange membranes (AEMs) have attracted considerable attention as next generation fuel cells, because they can use non-noble catalysts and various fuels. The SAFCs show complex water behaviors; water is generated at the anode and is reacted at the cathode. Hence, SAFCs using gas as the fuel show low cell performance due to anode flooding and membrane drying. Although control of water behavior is crucial to improve cell performance, there are many parameters to be considered such as AEM properties and operating conditions. In such situation, modeling approach is effective because of less time-consuming, inexpensive, and more versatile than experimental approaches.
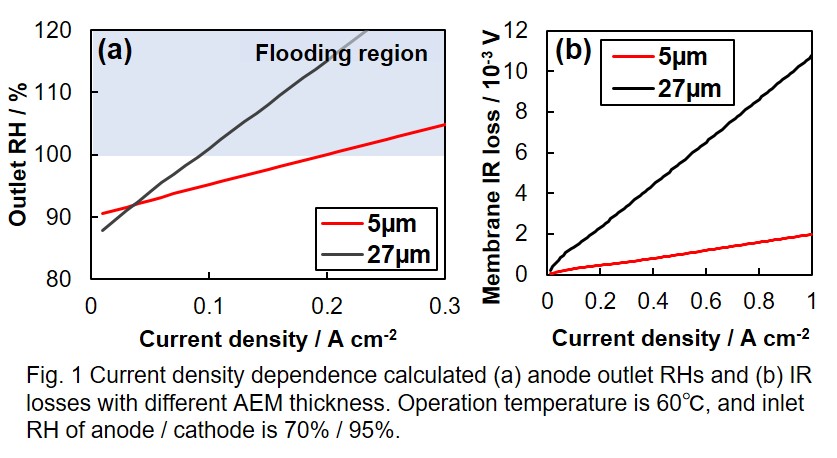
In this study, we make a mass balance model for SAFCs considering water transport in AEM and clarify guidelines to suppress anode flooding and membrane drying. Pore-filling anion conducting membrane[1] is used as AEM, and parameters needed for the calculation such as ion conductivity and water content of AEM were experimentally obtained. To make the AEM model, two mass balances of whole cell and AEM are considered. Then, outlet RHs and membrane IR loss were calculated by solving the two mass balances simultaneously under specific operating conditions and AEM thickness.
Fig. 1(a) and 1(b) show current density dependence of calculated outlet anode RHs and membrane IR losses with different AEM thickness, respectively. With thinner AEM, an increase in current density was obtained at 100% RH, which indicate the flooding is suppressed. Further, the suppression of membrane drying in the thinner AEM was confirmed by the low membrane IR loss. These results show that thinner AEM is effective for suppressing both anode flooding and membrane drying.
[1]Y. Oshiba et al., Journal of Power Sources , 345, 221-226 (2017)
Direct methanol fuel cell (DMFC) has attracted attention as an alternative power source because of high energy density of liquid methanol. One of the main issues for DMFC is the sluggish kinetics of methanol oxidation reaction (MOR) at the anode. Therefore, we focused on developing a highly active anode catalyst by using reduced graphene oxide (RGO) having a high specific surface-area and a high electric conductivity as a catalyst support. Although many studies using RGO have been conducted so far, the effect of the particle size of RGO on the catalytic activity has not been sufficiently investigated. In this study, the catalyst was prepared using RGO with different average particle size, i.e., 5 μm (RGOL) and 0.5 μm (RGOS) and the effect of particle size of RGO on the catalytic activity was investigated.
PtRu catalyst supported on RGOL or RGOS was prepared from a GO slurry by using a high-power ultrasonic crusher followed by adding polydopamine (PDA) as a dispersant and PtRu deposition by a chemical reduction method using NaBH4.
For the prepared catalysts, PtRu/RGOL and PtRu/RGOS, characterization was carried out using TEM, FE-SEM, XRD and EDX. Catalytic activity of the catalyst was evaluated by cyclic voltammetry in an aqueous solution with 0.5 M CH3OH and 0.5 M H2SO4. Electrochemical surface area (ECSA) for the catalyst was also evaluated by CO stripping method.
PtRu/RGOS showed about 1.6 times higher ECSA and about 2.1 times higher MOR mass activity compared to that of PtRu/RGOL. The higher performance of PtRu/RGOS was attributed to the superior structure of the catalyst layer for facilitated mass transfer and ease of fuel access. This result suggested that RGO with the small particle size is proper for the support of PtRu nanoparticles to increase the MOR activity of PtRu/RGO in DMFC.
The efficient conversion of energy is one of the urgent problems because of the sharp increase in current energy consumption. To meet the electricity demand, the development of fuel cells (FCs) have attracted great interest by their sustainable, renewable, efficient and eco-friendly electrochemical conversion of energy. For widespread commercialization of FCs, the main challenge is to reduce the cost of catalysts although expensive platinum-based catalysts have been applied. However, it is still challenging to develop an inexpensive and effective alternative catalyst, due to its sluggish reaction towards the oxygen reduction reaction (ORR).
As the alternative catalysts, transition metal and heteroatoms co-doped carbon material have been attracted by their low-cost and high performance. In particular, the carbon materials containing the bonding of cobalt and nitrogen (CoNx) acted as high-performance catalysts for ORR. However, the fine design of CoNx is difficult, due to the aggregation of metal. Herein, we present a new synthesis method of graphene (G) with highly-dispersed CoNx species as an excellent electrocatalysts, by using sequential treatments of mixing with sulfuric acid, Co2+ ion-exchanging and 2-methylimidazole coordination. And we also report its excellent electrocatalytic performance.
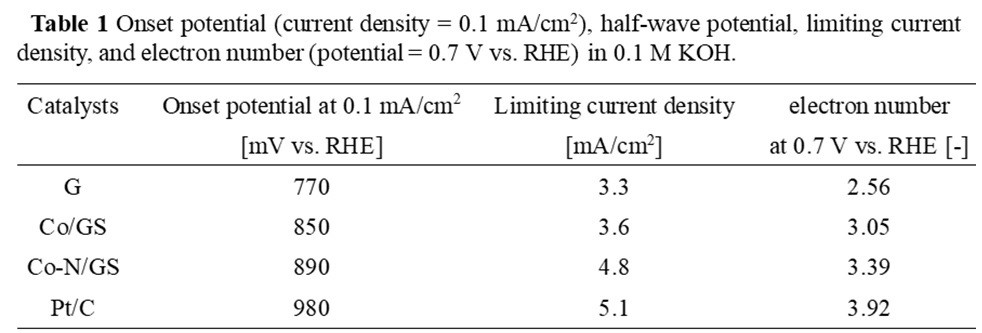
The highly dispersed cobalt species was observed on the graphene by TEM images and STEM images. The results of ORR measurements were shown in Table 1. The samples after the treatment of Co2+ ion-exchanging and 2-methylimidazole coordination were carbonized under N2 atmosphere and denoted as Co/GS and Co-N/GS, respectively. Obviously, G and Co/GS performed sluggish ORR, while Co-N/GS showed both excellent onset potential and limiting current density. In particular, the limiting current density is comparable to commercial platinum catalysts (Pt/C). Moreover, Co-N/GS exhibits a high current density, and the number of electrons (n) in the ORR are also higher than other samples.
Polymer electrolyte fuel cell (PEFC) is beginning to be put into practical use as residential and transportation power generation system. However, the cost of PEFC is still high, since expensive platinum (Pt) is used as the catalyst. Therefore, development of non-Pt catalyst is required. We have been studying silk-derived activated carbon (SAC) as non-Pt catalyst, which is inexpensive but fairly active compared to the conventional Pt catalyst. The structure of the electrodes fabricated from such catalyst should be designed with a different concept. In this study, we investigated the effect of catalyst layer thickness on the performance of PEFC using non-Pt catalyst.
Membrane electrode assembly (MEA) was prepared using SAC as cathode catalyst. Catalyst layers with different thicknesses were fabricated on a carbon paper and was hot pressed to a Nafion membrane. Terminal voltage vs. current density and electrochemical impedance spectra of each MEA were measured at 80 °C.
The iR-free polarization curves of each MEA were deconvoluted by fitting the activation overpotential. As the amount of catalyst was increased, the activation overpotential decreased and the remaining overpotential increased. It was assumed that this residual overpotential was not concentration overpotential, since the shape of the curve was not convex downward. The applied voltage dependence of the impedance spectrum was observed. As the applied voltage was increased, the interfacial resistance initially decreased and then became constant. Interfacial resistance independent of the applied voltage was assumed to be related to the proton conduction in the catalyst layer. The interfacial resistance related to the proton conduction in the catalyst layer increased as the amount of catalyst increased. Therefore, it is considered that the increase of the residual overpotential was due to the increase of the proton conduction resistance in the catalyst layer.
Graphene oxide (GO) membrane is expected to be an alternative electrolyte membrane having high proton-conductivity and less fuel-crossover for a fuel cell application. However, the proton-conductivity of GO membrane is still not enough. In this study, effect of an additional oxidation of GO and an addition of sulfonic acid group to GO on the proton conductivity were investigated.
A commercial GO slurry with 1.0 wt% GO was subsequently treated with NaNO3, KMNO4 and H2O2 to obtain the additionally oxidized GO (GOh). By adding vinyl sulfonic acid (VS) to the GO and GOh slurries, VS treated GO (GOVS) and VS treated GOh (GOhVS) membranes were prepared after filtering and drying. Membrane electrode assembly (MEA) was fabricated by sandwiching the membrane by carbon papers with Pt/C catalyst on both sides. Current-voltage performance of the MEA as a H2-O2 fuel cell was evaluated, and the proton conductivity of the membrane was calculated from the ohmic resistance assuming that the cell resistance was governed by that of the membrane.
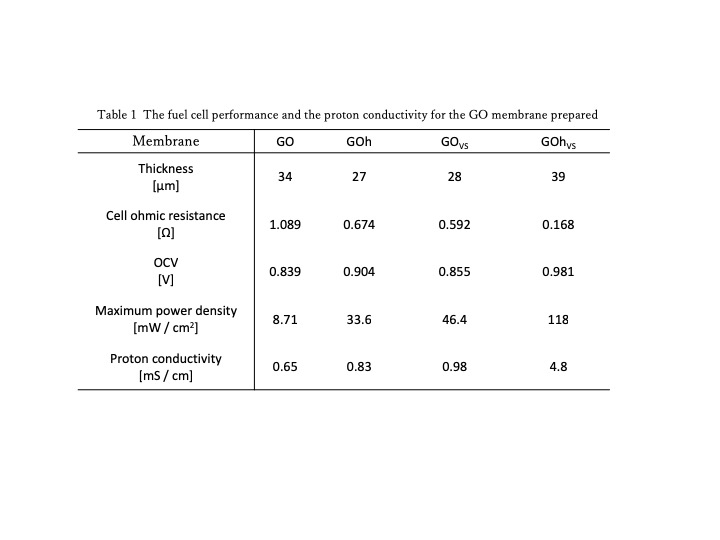
The proton conductivity of the membrane was higher in the order of GOhVS > GOVS > GOh > GO and found that each of the additional oxidation of GO and the VS treatment increased the proton conductivity as shown in Table 1. GOhVS membrane of which GO was additionally oxidized and also treated with VS showed the highest conductivity with 118 mW cm-2 fuel cell power-density. It was more than one order of magnitude higher than that of GO. It was found that the additional oxidation tof GO and the VS treatment are effective in improving the conductivity of GO membrane.
Polymer electrolyte water electrolysis (PEWE) is a key technology for the production of hydrogen in the power-to-gas process. As polymer electrolyte membranes for PEWE, perfluorosulfonic acid (PFSA) polymer membranes such as Nafion® are commonly used. In PEWE, although the produced hydrogen needs to be stored in pressurized gas tanks for transportation, the cell performance drops by high hydrogen crossover under the pressured condition. On the other hand, hydrocarbon-based polymer membranes including poly (arylene ether sulfone) (SPES) are expected to have a lower gas permeation property than the PFSA polymer membranes. However, the swelling of these hydrocarbon membranes is higher than the PFSA polymer membranes, which is a severe problem in fully humidified operating conditions of PEWE. The membrane with reduced swelling could be fabricated by a pore-filling technique, where the polymer electrolyte is filled inside pores of the porous substrate [1].
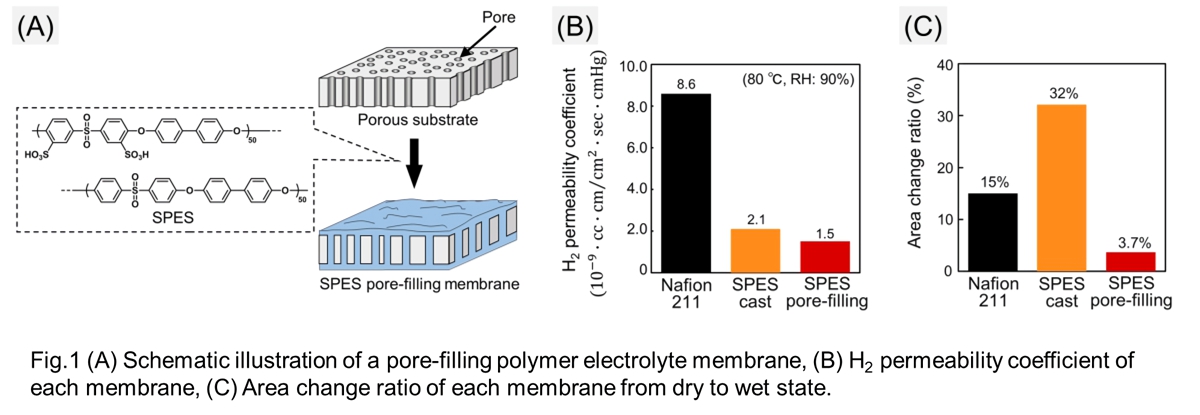
In our study, we prepared a pore-filling polymer electrolyte membrane using SPES for PEWE (Fig. 1A). This pore-filling membrane was prepared by drop casting 3 wt% SPES (IEC: 2.1 meq/g) solution over a 24 μm polyethylene porous substrate at 80 °C. The properties of the SPES pore-filling membrane was compared with that of SPES cast membrane and commercial Nafion 211.
The hydrogen permeability coefficient of the SPES cast membrane was found lower than Nafion 211, and that of the SPES pore-filling membrane is comparable with that of the SPES cast membrane (Fig. 1B). Moreover, the pore-filling membrane showed a lower swelling ratio between dry and wet conditions compared to the cast membrane (Fig. 1C). The results indicate that the SPES pore-filling membrane has both reduced hydrogen permeation and lowered swelling properties, which is suitable for PEWE applications.
[1] Nishimura, H. and T. Yamaguchi, Electrochemical and solid-state letters, 2004. 7(11): p. A385-A388.
Fuel cells combined with carbon-free hydrogen productions, such as water electrolysis, are the most promising energy strategy to mitigate the global warming and climate changes. Since the reactions for fuel cells (oxygen reduction reaction, ORR and hydrogen oxidation reaction, HOR) and water electrolysis (oxygen evolution reaction, OER and hydrogen evolution reaction, HER) are heterogeneous electrochemical reactions, they require catalysts to reduce the activation barriers during the electrochemical reactions. In addition, since the ordinary catalysts used in the systems are noble metal such as Pt, Ru, Ir, etc., development of cheap catalysts is another key issue in the fields. In this presentation, alloys of both noble and transition metals are fabricated by electrodeposition method. The composition and shape of the catalysts were controlled through the modification of the electrolyte composition and the deposition potential. For the ORR of fuel cells, PtM alloys were electrodeposited and their ORR characteristics through the modification of the electronic structures were studied. In the case of hydrogen-involving reactions such as HOR and HER, the relatively fast kinetics enables the possible use of cheap Ni-based alloys as the catalysts materials. Addition of small amounts of noble metals to Ni or control of the electrochemical surface area as well as the crystalline structures enhance the reaction kinetics close to those by Pt-based materials. A variety of combinations of Ni-based alloys was fabricated by electrodeposition and their characteristics on HOR and HER were investigated.
Energy issue has become a considerable topic because of the rapid population growth and industrialization of the world. To reduce carbon emission and environmental pollution from burning fossil fuels, the development of alternative energies is urgent and essential nowadays. Hydrogen has been regarded as one of the most promising energy carriers because of its high gravimetric energy density and clean byproduct, water, when in use as an energy source. Electrolytic water splitting driven by renewable energies such solar and wind energies is considered a most promising environmentally friendly hydrogen production technology, which splits water into hydrogen and oxygen without hazardous byproducts. The development of high-efficiency and noble-metal-free electrocatalysts is the most crucial and challenging part of electrolytic water splitting. In this study, we successfully grown Ni-Fe based bi-metallic metal-organic frameworks (MOF) on nickel foam, denoted as (Ni, Fe)(BDC-NH2)(DMF,F)/NF, for both oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) involved in alkaline electrolytic water splitting. The as prepared MOF loaded nickel foam exhibited outstanding electrolytic activities, achieving overpotentials of 286 and 348 mV for the OER and 250 and 353 mV for the HER at current densities of 50 and 500 mA cm-2, respectively in 1.0 M KOH, showing substantial activity enhancements as compared to pristine nickel foam. The OER efficiency enhancement may be attributed to the synergistic effects between Ni and Fe and the presence of fluoride, which is a strong electron withdrawing ion. As for the HER, the presence of the electron donating group, –NH2, in the organic linker is beneficial to the reduction power of the catalyst.
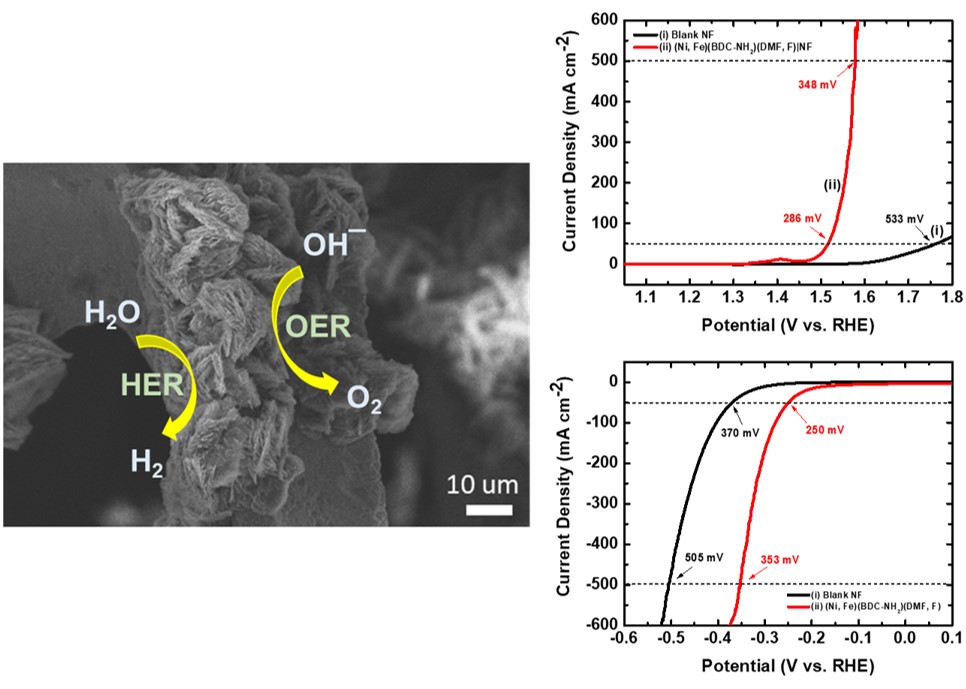
Efficient use of renewable energy resources is crucial to meet our future energy demand. Alkaline water splitting can produce hydrogen using electricity generated from intermittent natural energies, such as solar and wind. The oxygen evolution reaction (OER) is often considered as the bottleneck in water splitting because the OER still has large overpotential. RuO2 and IrO2 have high activities for OER, but these catalysts based on precious metals are very costly. In addition, high operating potential in OER region triggers corrosion of carbon supports of catalysts, leading their low durability.
Perovskite oxides with nonprecious metals have been recently attracting much interest because they are inexpensive and show high electrocatalytic activity. For electrochemical reaction, conductive supports need to be combined since most perovskite oxides have insufficient electrical conductivity. However, carbon supports are not suitable due to carbon corrosion.
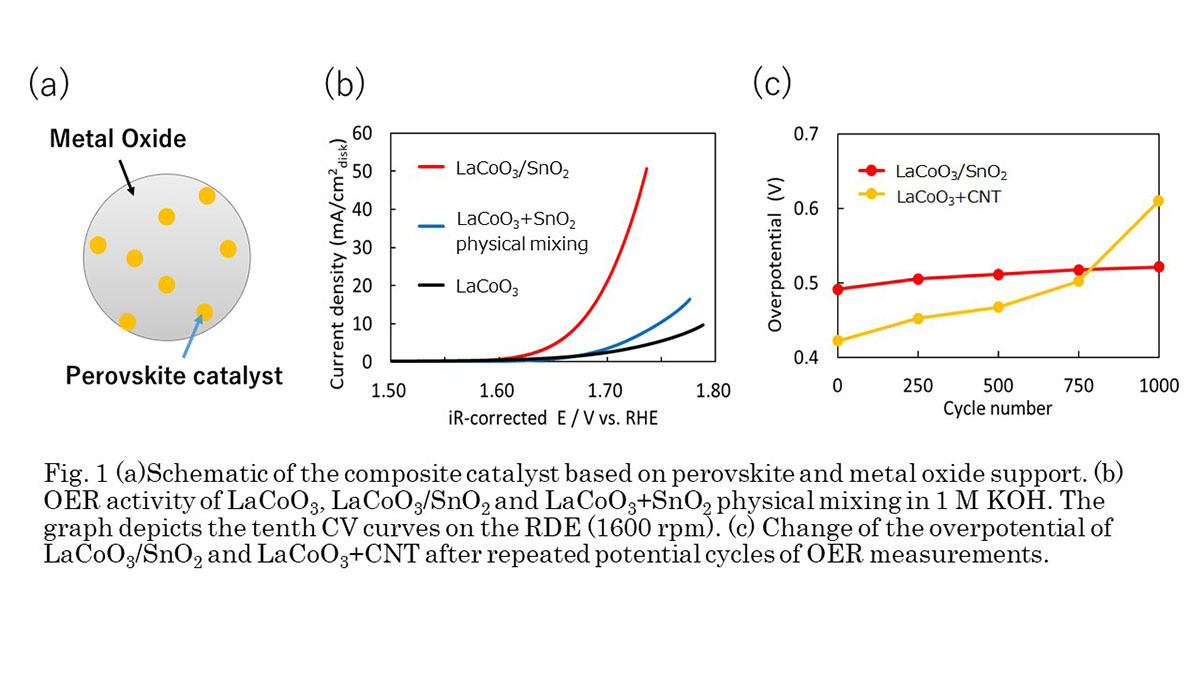
Herein, we develop a composite electrocatalyst composed of perovskite oxide as catalyst and conductive metal oxide as support as shown in Fig. 1(a), and we have chosen LaCoO3 and SnO2, respectively. LaCoO3 is a model perovskite compound, which is well-known as OER catalyst. SnO2 is electrical conductive oxide and possesses high thermal stability and alkaline stability, suggesting that SnO2 can be stable during the synthesis of LaCoO3 and measurements in alkaline solution. Fig. 1(b) shows OER activities of LaCoO3, LaCoO3/SnO2 and physically-mixed LaCoO3+SnO2 and demonstrates that LaCoO3/SnO2 exhibited higher current than others. It implies a good interfacial contact in LaCoO3/SnO2. Moreover, Fig. 1(c) proves higher durability of LaCoO3/SnO2 than that of LaCoO3/CNT which employed carbon as support. These findings indicated that the composite electrocatalyst based on perovskite and conductive metal oxide support is a promising platform for development of highly durable OER catalysts.
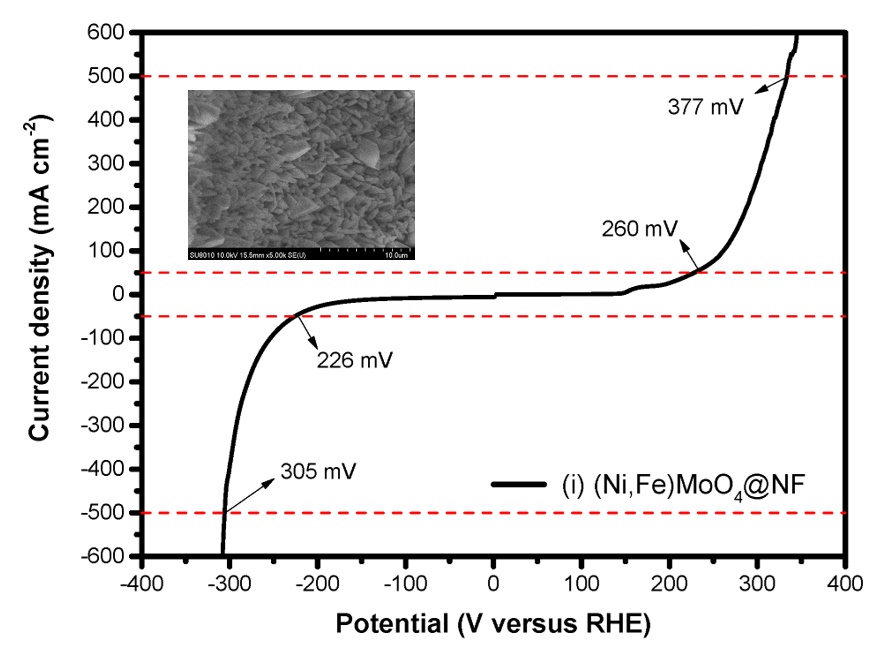
The development of green energy are getting increasing research attention. Hydrogen is an excellent energy carrier and is also considered an energy source suitable to serve as an intermediate energy carrier for solar and wind energies. The development of highly efficient, stable, and low-cost electrocatalysts to reduce the reaction potential of electrolytic water splitting can facilitate conversion of green energies into hydrogen energy. In this study, we successfully grew Ni-Fe-Mo based nanorod arrays on nickel foam, denoted as (Ni, Fe)MoO4@NF, as bifunctional electrocatalysts for overall water splitting. The product catalysts exhibited excellent electrocatalytic activities toward the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) in 1.0 M KOH, achieving overpotentials of 128, 226, and 305 mV at 10, 50, and 500 mA cm-2, respectively for the HER and overpotentials of 227 and 333 mV at 50 and 500 mA cm-2, respectively for the OER. The enhancement in OER efficiencies can be mainly attributed to the synergistic effects between Ni and Fe. And the enhancement in HER efficiencies can be mainly attributed to the synergistic effects between Ni and Mo. Furthermore, the nanorod array structure on nickel foam enlarges the reaction surface areas and thus the active site number for the water splitting reaction.
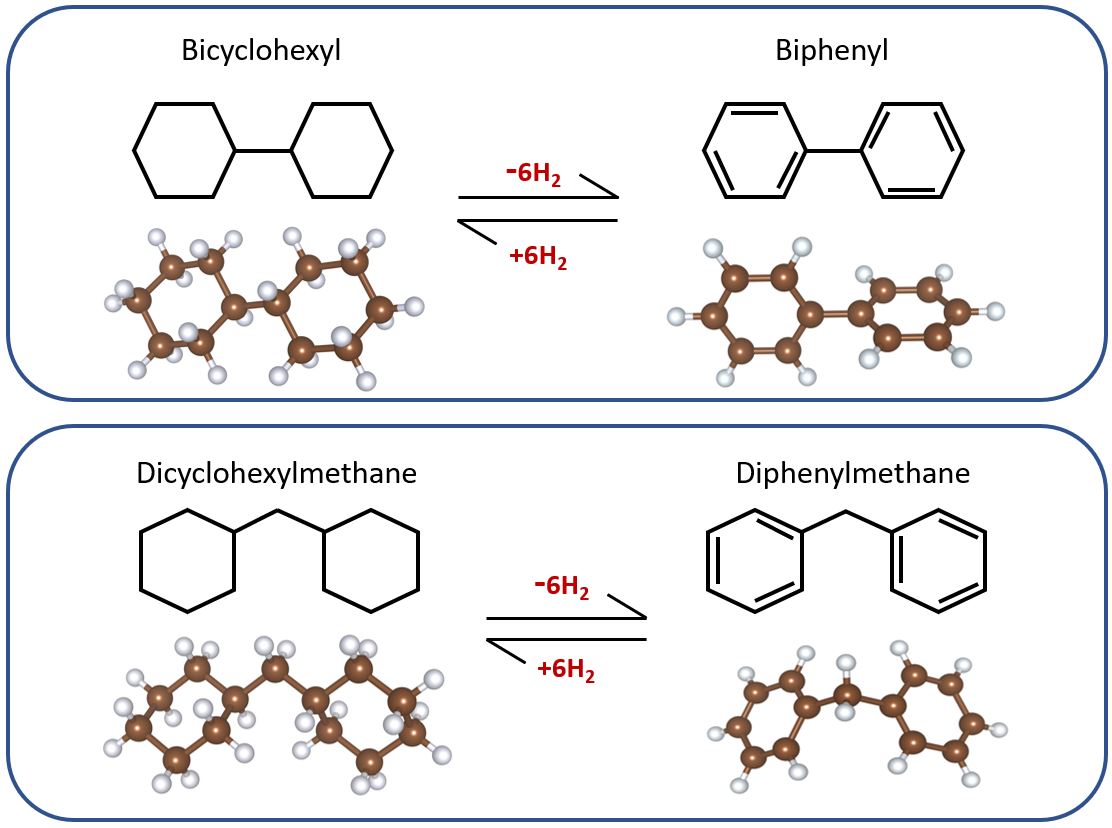
Hydrogen energy, an eco-friendly energy source for replacing fossil fuels, is widely regarded as a next-generation energy source with highly versatile application. However, there are technical limitations on hydrogen storage and production that should be satisfied for aspects of economy and efficiency. Among many technologies for solving the problems, liquid organic hydrogen carriers (LOHC) technology has been receiving much attention since it can be stored for a long time and safely transported to a destination using existing gasoline infrastructure. Recently, it has been experimentally reported that when biphenyl was mixed with diphenylmethane, it showed a reversible hydrogen storage and production capacity of 6.9 wt% H2 and 60 gH2/L, existing in a liquid state at room temperature. To understand this mechanism in detail, density functional theory (DFT) calculations were conducted using Gaussian 16 software package. We analyzed the reversible dehydrogenation reaction mechanisms based on the dehydrogenation enthalpy and free energy calculations. Furthermore, the same calculations were conducted with a various functional groups to propose higher efficient LOHC materials, which would make a great contribution to the development of optimal LOHC materials for the reversible hydrogen storage and production system.
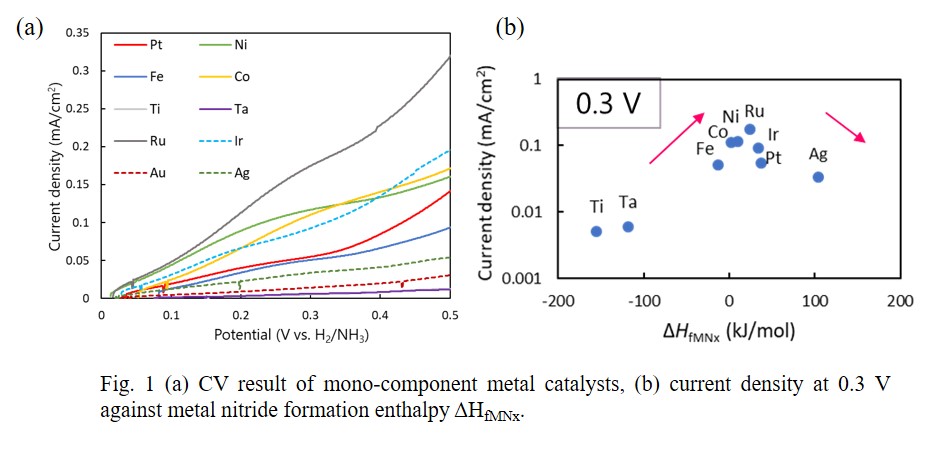
Hydrogen is a secondary energy source and can generate electricity by fuel cells without CO2 emission. Ammonia is considered for one of the promising hydrogen storage media. Ammonia has high gravimetric and volumetric hydrogen capacities (17.8 mass% and 107.3 kgH2/m3). And ammonia is easily liquefied at 1.0 MPa at room temperature. Hydrogen can be obtained by electrolysis of liquid ammonia. The theoretical standard voltage is 0.077 V, however the actual operation voltage is much higher due to the large overpotential for the anode reaction. In the anode reaction of N2 formation from NH2-, it is considered that either NH2- adsorption or N2 desorption can be the limiting step. Therefore, the catalyst effect on anode reaction was investigated by focusing on the metal nitride formation enthalpy ΔHfMNx which corresponds to the strength of metal-nitrogen bond. The electrolysis of liquid ammonia was performed using a three-electrode system with a counter electrode of a Pt plate, a reference electrode of a Pt wire (-2.59 V vs. SHE) and a working electrode of a catalyst plate (Ti, Ta, Fe, Co, Ni, Ru, Ir, Pt, Au, or Ag). The cyclic voltammetry (CV) of mono-component catalysts showed different activities for different metals (Fig. 1a). By plotting the current density at 0.3 V against the ΔHfMNx, the volcano plot was obtained (Fig. 1b). Next, to increase the catalytic performance, binary metal catalyst of Fe and Pt (Fe-Pt) having different bond strengths with nitrogen was prepared. Fe-Pt showed higher current density than the mono-Fe and mono-Pt catalysts at a low potential of 0.3 V.
NH3 is one of the key chemicals in the world. For example, NH3-based chemicals occupy the vital position of a fertilizer, which directly affects the world's food supply. Besides, NH3 is considered a promising H2 carrier because of its high energy density, easy liquefaction, and no CO2 emission during H2 evolution reaction. Today, NH3 is synthesized from syngas and N2 at high pressure (15-30 MPa) and at high temperature (500 °C), which is known as a Haber-Bosch process. If this synthesis is achieved without using fossil fuel, we will obtain carbon-free NH3. We focused on electrochemical synthesis of NH3 which can be operated at atmospheric pressure by electricity from renewable energy resources. We chose a solid phosphate electrolyte (a CsH2PO4/SiP2O7 composite) which conducts protons at around 200 °C, based on the studies of intermediate-temperature fuel cells. Both electrodes consisted of Pt on carbon with 1 mg_Pt cm-2. Ideally, the electrochemical synthesis proceeds as follows. At an anode side, H2 is separated to protons and electrons (H2 → 2H+ + 2e-). At a cathode side, N2 reacts with protons and electrons (N2 + 6H+ + 6e- → 2NH3). Using the electrolyte and electrodes, however, NH3 was detected not only from cathode but also from anode probably because of the NH3 leakage from the cathode to the anode and desorption/decomposition of NH3-like contaminations. Thus, we struggled with changes in experimental conditions and improvement in the apparatus and experimental procedure. Ag-Pd membrane was found to be effective for preventing the gas leak, and by inserting Ag-Pd membrane between anode and electrolyte, NH3 detection in the anode side was limited, and electrochemical NH3 synthesis was confirmed in the cathode side.
Ammonia has been paid attention as a hydrogen medium for hydrogen energy. The aqueous ammonia solution has a high hydrogen mass density (6.1 mass%) and a vapor pressure (0.08 Pa) lower than atmospheric pressure at 20 °C. Electrolysis is one of the methods of generating hydrogen from aqueous ammonia solution. Hydrogen and nitrogen are generated at the anode and cathode, respectively, and the theoretical decomposition voltage is 0.06 V. Platinum is often used for the electrode catalyst but has a problem of the large overvoltage of the anode.
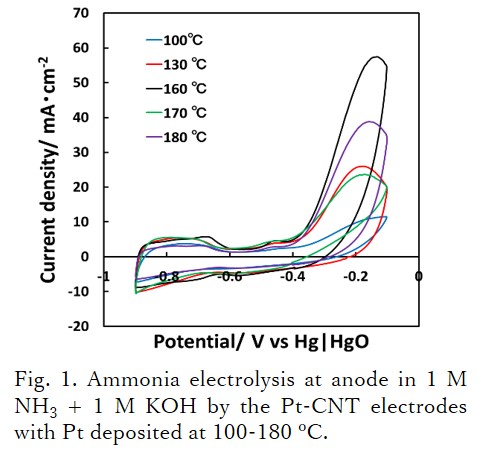
In this study, an electrode with platinum nanoparticles supported on carbon nanotube (Pt-CNT) was fabricated. CNT is a carbon material possessing a high specific surface area, that forms a sponge-like film by a dispersion-filtration process. The Pt-CNT electrode was adapted to electrolysis of ammonia aqueous solution to reduce the anode overpotential by increasing the Pt surface area.
Pt nanoparticles were deposited on CNT by polyol method at various temperatures of 100-180 °C. The electrochemical surface area of Pt was calculated from the amount of hydrogen adsorption and desorption in 0.5 M H2SO4 aq. The largest platinum surface area, 268 cm2 per 1 cm2 of electrode, was obtained for the Pt-CNT with Pt deposited at 160 °C. The ammonia electrolysis results at the anode with 1 M NH3 + 1 M KOH aqueous solution are shown in Fig. 1. The Pt-CNT electrode prepared at 160 °C and having the largest platinum surface area showed the highest peak current density and the lowest potential at 10 mA cm-2.
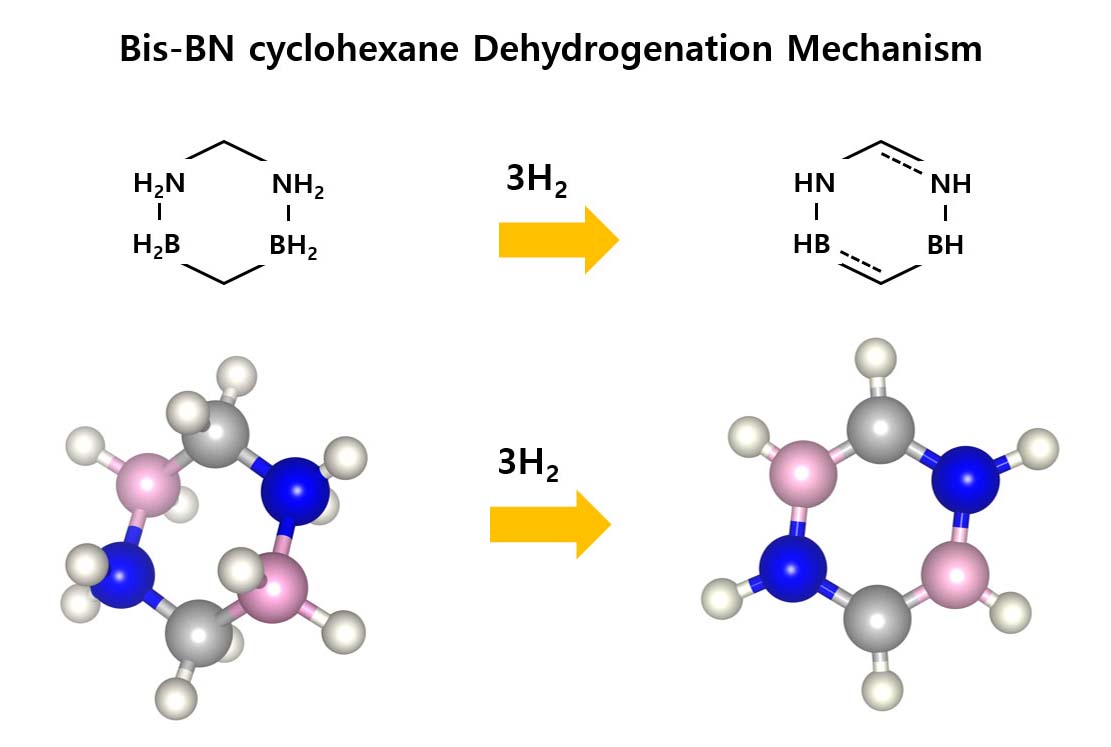
Among many eco-friendly energy sources, hydrogen energy has been attracting attention due to a higher versatility, but there is a difficulty in the practical use due to technical limitations in storing and transporting hydrogen. Liquid organic hydrogen carriers (LOHC) technology is one of the most promising alternative methods, storing hydrogen in a liquid state compound, thereby enabling stable and high-density hydrogen storage. Previou studies have reported various LOHCs such as metylcyclohexane, dibenzyl toluene, N ethylcarbazole, ammonia borane and bis-BN cyclohexane. Among them, it has been reported that bis-BN cyclohexane, a derivative of AB, can overcome the disadvantage of ammonia borane (i.e., AB is rapidly decomposed below 150 °C), because it is thermodynamically stable at 150 °C and has a hydrogen storage capacity of 4.7 wt%. In the current study, we conducted an ab-initio study to develop hydrogen storage materials with higher efficiency and reversibility using Gaussian 16 software package. The dehydrogenation mechanism of bis-BN cyclohexane was investigated by calculating the reaction enthalpies and free energies, and then highly efficient materials were screened by conducting the same calculation depending on various functional groups such as F, Cl, and CH3. When F was replaced in one of the H atom of bis-BN cyclohexane, the enthalpy and free energy for the dehydrogenation were 26 kcal/mol and 0.1 kcal/mol, respectively, which proved to be enhanced reversibility of bis-BN cyclohexane-based LOHC.
Electronic power generation in Japan contributed by thermal power generation in terms of economics, environmental compatibility, and stable supply. However, there is a problem that the environmental negative influence is high, such as the formation of greenhouse gases emitted when burning fossil fuels. Therefore, hydrogen has recently been attracted attention as a power source. Hydrogen has an advantage compared to the other energy medias, i.e., hydrogen can use be produced from various primary energies including renewable energies, and electricity is obtained by using a fuel cell. However, there is also an issue that the cost is high for transportation and storage due to the use of a high pressure tank because of the low volumetric energy density. Therefore, we propose an electrochemical device for production of hydrogen from methylcyclohexane, which is an energy carrier. The reaction, separation, and compression processes are performed by one device, the cell structure is similar to a PEFC, and the reaction principle is the electrolysis of methylcyclohexane by an external voltage.The formation of proton from methylcyclohexane carries out at an anode catalyst (reaction process), separation between hydrogen and organic compound by the Nafion membrane (separation process), and electrochemical compression based on the Nernst equation (compression process). In this study, hydrogen was obtained from reaction, separation, and compression from pure methylcyclohexane, and the concentrations of hydrogen and impurities were evaluated by gas chromatography. As a result, a current density was more than 50 mA cm–2 at a cell voltage of –2.0 V.
As an eco-friendly energy source to replace fossil fuels, hydrogen energy is seen as a next-generation energy source with its high general purpose applicable. However, there are technical limitations to stable hydrogen storage and release for economic feasibility and efficiency. Liquid organic hydrogen carriers (LOHCs) can be one of the ways to overcome this. In previous studies, it was demonstrated experimentally that biphenyl exists in a liquid state at room temperature when mixed with diphenylmethane, and has a hydrogen storage capacity of 6.9 %wtH2 and 60 g/L. Our preliminary study demonstrated that the dehydrogenation efficiency of biphenyl has increased when carbons were replaced with boron (B) and nitrogen (N) in the molecule. Based on the findings, the current study investigated the reversible dehydrogenation and hydrogenation mechanisms of BN-biphenyl and BN-diphenylmethane by predicting reaction enthalpies ($Delta;H) and free energies ($Delta;G) based on density function theory (DFT) calculations. In addition, alternative materials with higher efficiency are to be screened by adding functional groups (F, Cl, NH2, etc.) in the molecules. This will contribute to the development of an optimal hydrogen storage and release of highly efficient LOHCs.
Hydrogen, which is clean and storable fuel, is an attractive energy carrier for energy and environmental issues. In addition, hydrogen plays a vital role in chemical industry as a reactant. Nowadays, hydrogen is mainly produced from fossil fuels by steam reforming with CO2 emissions, so new hydrogen production process which is eco-friendly has been desired. Thermocatalytic decomposition of hydrocarbon is attracting attention because there is no emission of greenhouse gases such as CO2 during hydrogen production. Several researchers have investigated various catalysts for this reaction, but there is a problem of carbon deposition on the surface which blocks active sites of the catalyst. Based on the situation, “chemical looping methane cracking system” has been proposed. This system is composed of two reactors. In a first reactor, methane is cracked into H2 and carbon, and in a second reactor, deposited carbon is oxidized by CO2 into CO. The system has advantages in terms of separated syngas production and CO2 utilization. As an oxygen carrier, Fe/BaZr0.9Y0.1O3 composite was reported in our previous study, which exhibited high reaction kinetics for methane cracking because of high proton conductivity of BaZr0.9Y0.1O3
In this study, we synthesized Fe-doped BaZr0.9Y0.1O3 and investigated the reaction activity of these catalysts by thermal gravimetric analysis and gas analysis. Fe-supporting method, i.e., Fe doping, and the amount of Fe influenced the states of deposited carbon in methane cracking. For example, direction of growth and diameter of carbon fiber, and reaction kinetics also changed by the oxygen carriers. On the other hand, the carbon oxidation reaction with CO2 completed in a relatively short time for each catalyst. Besides, cyclic stability was investigated. Through the kinetic measurements, we discussed the effect of the feature of prepared catalysts on reaction kinetics and the efficiency of the system.
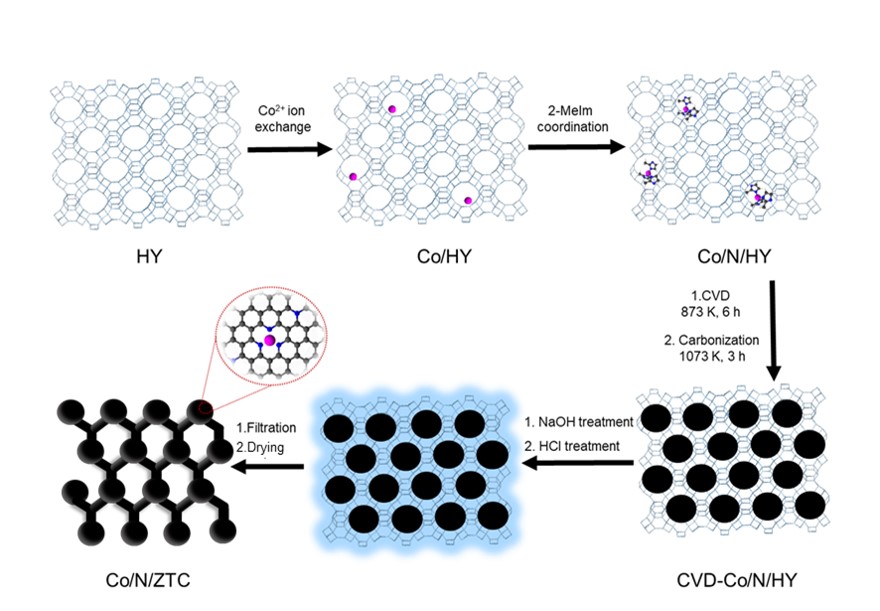
Zeolite templated carbons (ZTCs) composed of curved and single-layer graphene frameworks have uniform micropores (ca. 1.2 nm) and high surface areas (~4000 m2 g-1). Due to their outstanding properties originating from the porous structures, ZTCs have been utilized in many applications, e.g. hydrogen storage, CO2 capture, catalysts and electrochemical capacitors. To expand the utilization of ZTCs, metal atoms or heteroatoms have been doped to ZTCs. However, it is still challenging to fabricate single atomic transition metals coordinated with heteroatoms in ZTCs. In this work, we synthesized Co/N doped ZTC (Co/N-ZTC) by complexing Co ion with 2-methylimidazole in Y zeolite to expand the further utilization of ZTCs as shown in Figure 1. The first step is to anchor Co/2-methylimizole complex into HY zeolite as a precursor of Co/N-ZTC by complexing Co ion with 2-methylimidazole in HY zeolite. The next step is chemical vapor deposition (CVD) using methanol as a carbon source, followed by carbonization process. The last step is base and acid treatments to remove HY zeolite and aggregated Co particles. The obtained Co/N-ZTC has a high surface area (ca. 2000 m2 g-1) and single atomic Co species in CoNx moieties, which actually contributes to its excellent catalytic performance on oxygen reduction reaction. This synthetic concept is beneficial to fabricate single atomic transition metals coordinated with heteroatoms in ZTCs.
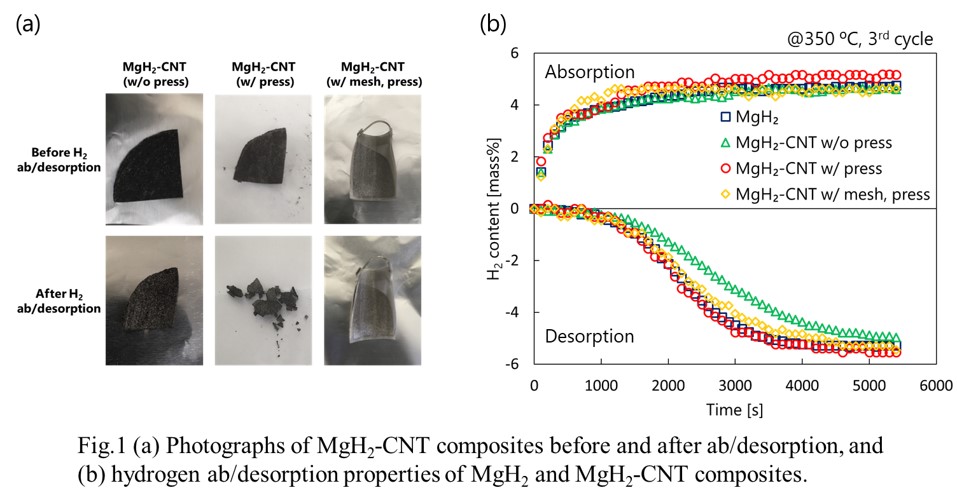
Hydrogen is paid attention as clean energy because it does not emit CO2 when converted to electricity. MgH2 is one of the promising hydrogen storage materials because of its high volumetric and gravimetric hydrogen densities and abundance. However, Mg expands and shrinks during hydrogen absorption and desorption, respectively. MgH2 powder pulverizes during hydrogen absorption and desorption cycles. In the tank filled with MgH2, pulverized powder drops and the density of packed bed at the bottom increases. This causes a problem of tank deformation during cycles. In this research, in order to absorb volume change of MgH2 and fix pulverized powder during cycles, MgH2 particles were retained in carbon nanotubes film. MgH2 and carbon nanotubes were co-dispersed in N-methyl-2-pyrrolidone. The MgH2-CNT composite was synthesized by vacuum filtration of the dispersion. The structure of MgH2-CNT composite (MgH2-CNT w/o press) was maintained after hydrogen ab/desorption (Fig.1 a), however, the hydrogen desorption rate was lower than that of MgH2 powder (Fig.1 b). In order to improve desorption rate, the MgH2-CNT composite was pressed to decrease its porosity. As the result, the desorption rate of pressed MgH2-CNT composite (MgH2-CNT w/ press) was improved compared to MgH2-CNT w/o press, but the film structure was broken into pieces. The MgH2-CNT composite was wrapped in stainless mesh and pressed (MgH2-CNT w/ mesh, press) to support the film structure. The composite achieved improved desorption rate and maintained film structure during hydrogen ab/desorption cycles.