
Glass transition temperature (Tg) of pharmaceutical polymers could be depressed by the pressurization with CO2. This phenomenon could be utilized for the formulation of pharmaceutical polymers in a short time and at mild temperature. However, there are some problems of the safety and cost under high pressure. Addition of organic solvent in CO2 could depress the Tg of polymers. Organic solvent is harmful to human, so addition of small amount of harmless solid co-solvent might be useful to lower the pressure. In this study, effects of solid co-solvent on the Tg of pharmaceutical excipients were examined with a visual method using the transmitted light intensity.
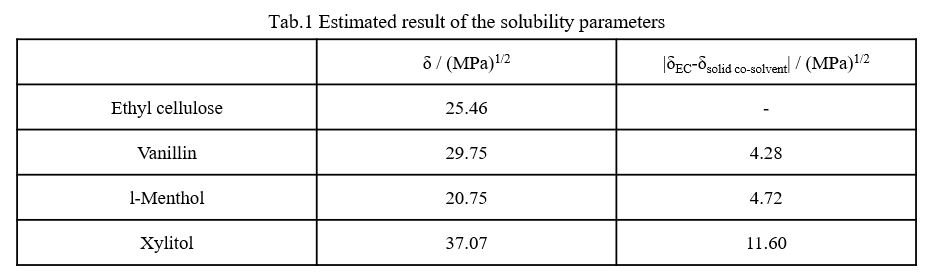
The Tg of Ethyl cellulose (EC)/CO2/ solid co-solvent systems was measured. For example, Tg of EC under atmospheric pressure was depressed by physical mixing with Vanillin (VAN) or l-Menthol (MEN) as solid co-solvent. Tg of EC with VAN or MEN wasn't depressed much under CO2. On the other hand, Tg of EC wasn't depressed by physical mixing of Xylitol (XYL). Therefore, it could be concluded that there are combinations of EC and solid co-solvent which can depress the Tg of EC and the Tg could be depressed by only addition of suitable solid co-solvent without CO2 pressure.
The state transition of EC with solid co-solvent was easily observed visually. It could be considered that Tg of EC was depressed by melted VAN or MEN dissolving into EC and wasn't depressed by melted XYL. From these facts, affinity between EC and solid co-solvent was examined by the solubility parameter (δ) which is an index of affinity between substances. The solubility parameters estimated by the group contribution method reported by Fedors [1] successfully explained that the affinity of polymers and solid cosolvent controls the Tg depression.
References
[1] Robert F. Fedors, Polymer Engineering and Science, Vol. 14, No. 2, California, (1974)
Biomass is a renewable and abundant resource that can reduce our dependence on the dwindling fossil fuel reserves. In particular, sugars are simple molecules that can be converted to biofuels and a wide array of platform chemicals. As found in plants and other organisms, fermentable sugars commonly exist as the repeating units in polysaccharides, which further form a complex network among themselves. Presently, one of the major challenges is to extract these sugars from these networks in an efficient and cost-effective way. The cleavage of the glycosidic bonds present in the polysaccharide structure is an important step in the process and understanding its mechanism can provide the insights necessary to develop better technologies to obtain the sugar monomers.
In this study, we used quantum calculations to determine a mechanism of the production of sugars from the acid-catalyzed hydrolysis of two model compounds, namely, cellulose and fucoidan. Glucose is the monosaccharide produced from the former and fucose from the latter. In both cases, our simulations showed that the process involves four steps. First, the oxygen in the glycosidic bond is protonated by an acid. Then, the C–O bond is broken, yielding a carbocation of either glucose or fucose. Next, the oxygen of a water molecule bonds with the positively charged carbon in the intermediate structure. Finally, the molecule is deprotonated, yielding the desired sugar monomer. Consistent for both cellulose and fucoidan, the initial step (i.e., protonation) is the rate-determining step because it requires the highest activation energy. Furthermore, our calculations showed that the depolymerization of cellulose via acid-catalyzed hydrolysis is less energy demanding than that of fucoidan.
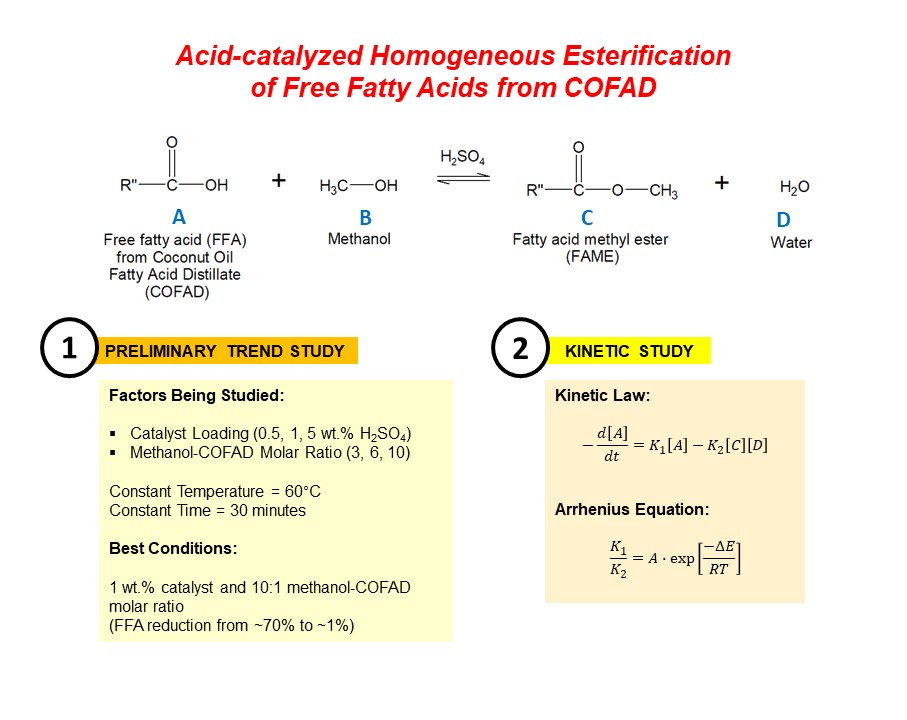
Because of the insufficient supply of refined coconut oil as a raw material for biodiesel production in the Philippines, an exploration on the potential use of oil refining by-products, particularly coconut oil fatty acid distillate (COFAD), as an alternative feedstock has been done. The main problem encountered in the use of COFAD is its high free fatty acid (FFA) content. A pretreatment step via esterification using methanol as a solvent is needed in order to reduce the FFAs present by converting it to fatty acid methyl esters (FAME). In this study, the conversion of FFA in COFAD to FAME was done via homogeneous esterification using sulfuric acid as catalyst. The effect of catalyst loading (0.5, 1, 5 wt.% sulfuric acid) and methanol-to-COFAD molar ratio (3, 6, 10) were studied first in order to determine the best process conditions that would lead to a maximum FFA conversion and FAME content. It was found that a low catalyst amount (1 wt.%) and a high methanol-to-COFAD molar ratio (10:1) at a time of 30 minutes is a favorable combination to reduce the FFA from ~70% to ~1%. A kinetic study was further done using the best process conditions for esterification and the results were found to fit in the kinetic model having a pseudo-homogeneous first-order in the forward direction and second-order in the reverse direction. The effect of temperature on the rate of reaction was also determined by fitting the kinetic data to the Arrhenius Equation. The results of this study suggest that COFAD can be a suitable alternative feedstock for the production of biodiesel in the Philippines.
Polycarbonates (PCs) have excellent features such as transparency, impact resistance, thermal stability, dimensional stability, and flame resistance. A non-phosgene process for PC production from carbon dioxide, ethylene oxide, and bisphenol A, has been developed. This process provides an effective use for carbon dioxide, which is a greenhouse gas and an abundant carbon resource, and has attracted much attention in terms of greener and more sustainable chemistry. Diphenyl carbonate (DPC) is an important intermediate material in these non-phosgene processes. The DPC purity directly affects the downstream PC quality. High-purity DPC is important in the production of PC for optical media applications. One of the options for DPC purification is crystallization. Knowledge of solid–liquid equilibrium (SLE) data is crucial in the design and development of crystallization processes. Our group determined the SLE of several binary systems containing DPC. However, more experimental SLE data for systems containing DPC are still needed.
The object of this work was to obtain SLE data for ternary systems containing DPC. We investigated the SLE of two ternary systems, i.e., methanol + DMC + DPC and phenol + DMC + DPC in the reaction involved in the non-phosgene PC process. Two ternary SLE data were experimentally determined by a synthetic, visual technique designed in our laboratory. In the measurements of ternary systems, DPC free basis mole fraction of methanol or phenol “α” was changed from 0.3 to 0.9, and changes of SLE behaviors were discussed with an increase of α. The SLE predictions for two ternary systems were compared with the experimental SLE data on the basis of the NRTL parameters obtained from the constituent binary SLE data. Also, the National Institute of Standards and Technology (NIST)-modified universal functional activity coefficient (UNIFAC) group contribution model was also used for the prediction of two ternary systems.
Direct oxidation of formic acid is prominent in fuel cells. We recently reported the catalytic
activity of LaCoO3 for this reaction [Electrochem. Commun., 99, 1-4, 2019]. For the first time, density functional theory (DFT) calculations are used for analyzing the mechanism of this reaction. We sampled a different number of HCOOH adsorption configurations as adsorption plays a crucial role in oxidation. Similarly, proton adsorption in different oxygen environments of the surface was analyzed. By considering the electro-oxidation environment of HCOOH, comparison of decomposition was done with and without water. This study is expected to aid the understanding oxidation of HCOOH on noble-metal free material LaCoO3.
Graphitic carbon nitride, whose activity is due to the electronegativity difference between the carbon and nitrogen atoms, has gained popularity among metal-free catalysts. Experiments with graphitic carbon nitride have shown that addition of a conductive carbon support to GCN improves its activity towards oxygen reduction reaction (ORR) 1, 2. Following previous studies on doped GCN, substitution of nitrogen or carbon with heteroatoms such as sulfur and phosphorus can further enhance GCN's ORR activity 3, 4. In this work, we investigate through a density functional theory-based calculation the effect of substitutionally doping sulfur and phosphorus on the graphitic carbon nitride/graphene layer in terms of adsorption energies and charge transfer extent upon oxygen adsorption. The results of the calculations suggest that sulfur doping provides higher adsorption energy compared to phosphorus doping. In terms of doping location, the calculations reveal that doping along the edge sites gives the most energetically favorable structure for oxygen adsorption. Moreover, this work considers the possible relationship between the oxygen-GCN/graphene separation distance and the oxygen bond length as an indicator of the interaction of molecular oxygen as it adsorbs onto the GCN/graphene surface.
1 S. Lyth et al. J. Phys. Chem. C 113, 47, 20148-20151 (2009).
2 S. Yang et al. Angew. Chem. Int. Ed 50, 23, 5339-5343 (2011)
3 Y. Zheng et al. J. Am. Chem. Soc. 133 50, 20116-20119 (2011)
4 Q. Han et al. J. Mater. Horiz 4, 832-850 (2017)
