
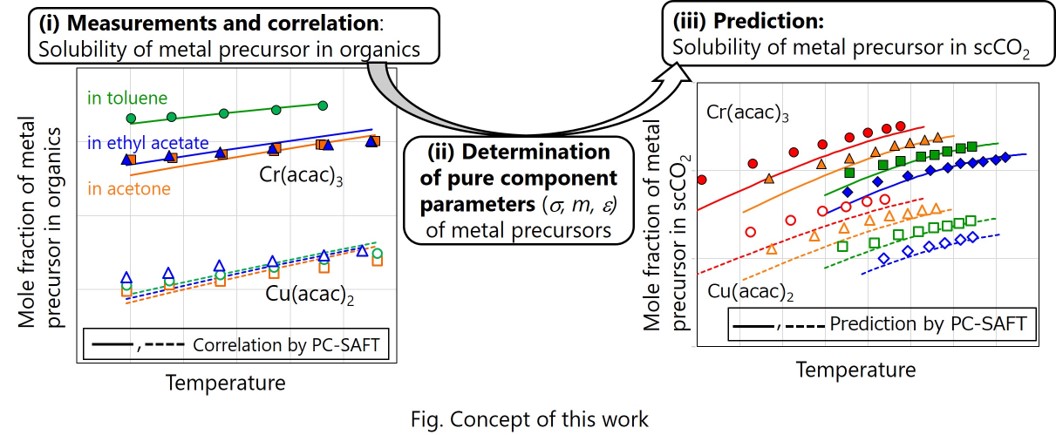
Deposition of metal particles into porous supports such as mesoporous silica using supercritical carbon dioxide (scCO2) has attracted much attention to prepare supported catalyst because of its ability to disperse metal particles into pore structures [1, 2]. Prediction methodology of the solubility of metal precursors in scCO2 is important to efficiently design the supported catalyst using the scCO2 deposition method.
In this work, the prediction of solubilities of various acetylacetonate-type metal precursors in supercritical carbon dioxide was performed using a thermodynamic model with the PC-SAFT (perturbed-chain statistical associating fluid theory) equation of state [3]. Pure component parameters for the metal precursors (segment number, segment diameter, and dispersion energy) were determined with correlations to solubility data of the metal precursors in various organic solvents, which were newly measured in this study. The pure component parameters of the metal precursors determined with the correlations were applied to predict literature data of solubilities of the metal precursors in scCO2 [4, 5] using the PC-SAFT equation of state. The PC-SAFT equation of state could reproduce the solubilities over wide temperature and pressure ranges for various precursors under almost all conditions without kij.
References
[1] J. Morere et al., RSC Adv., 5 (2015) 38880-38891.
[2] S.B. Barim et al., Microporous Mesoporous Mater., 245 (2017) 94-103.
[3] J. Gross, G. Sadowski, Ind. Eng. Chem. Res., 40 (2001) 1244-1260.
[4] M. Haruki et al., Fluid Phase Equilib., 297 (2010) 155-161.
[5] M. Haruki et al., J. Chem. Eng. Data, 56 (2011) 2230-2235.
Alanine condensation in high temperature and high pressure water were performed by molecular simulation and flow-type experiment to investigate catalytic role and solvation effect of water. Relative energies of the species consisting alanine condensation and corresponding transition structures were optimized by geometry optimization with B3LYP/6-31+G(d,p) level of theory. We considered four selected reaction mechanisms of alanine condensations such as anionic, zwitterionic, cationic, and non-ionic form. We also calculated a water molecule participation onto non-ionic reaction mechanisms to quantify the catalytic role of water. Activation energies considered reaction mechanism were ranged 117 kJ/mol to 190 kJ/mol, while alanine condensation by zwitterionic mechanism showed a lowest activation energy. Activation energies in solution were calculated by the sum of the activation energy from DFT calculation and the solvation free energy from Monte Carlo simulation with the theory of energy representations by Matubayasi and Takahashi. Solvation free energy difference between reactant and transition state indicated that the alanine condensation with zwitterionic mechanism was promoted by high temperature and high pressure water, on the other hand, the alanine condensation by anionic mechanism was inhibited by water as reaction media. This solvation effects by solvation free energy difference did not have great influence on activation energy due to the solvation effect contributed around 10% to activation energy. The temperature dependence of alanine condensation by means of the flow-type experiments was indicated the second-order reaction and the activation energy was 48.5 kJ/mol. Future investigations on ionic mechanism of alanine condensation are needed to elucidate the catalytic role of water molecule and solvation effect of water as reaction media. The solvation effect of high temperature and high pressure water on alanine condensation should be responsible for the preferential solvation around reaction species, which would be shown by the analysis of solution structure in the presentation.
We developed non-polarizable force field for ionic liquids by using first-principles calculations based on density functional theory(DFT). The cation is 1-alkyl-3-methyl-imidazolium, N-metyl-N-proylpyrrolidinium, and N-butyl-N,N,N-trimetylammonium. The counter anions are chloride, tetrafluoroborate, bis(fluorosulfonyl)amide, and bis(trifluorosulfonyl)amide. The DFT calculations were implemented with the QUICKSTEP of CP2K[1]. The PBE-type generalized gradient approximation was employed for the exchange-correlation functional. The atomic charges assigned in the nonpolarizable force field were evaluated with the Blöchl method[2]. The initial van der Waals parameters were set to OPLS model developed by Lopes et al.[3], and they were optimized to reproduce the intermolecular forces obtained with DFT calculation. We performed the molecular dynamics calculations in the framework of the optimized molecular force fields, and evaluated transport coefficients with Green-Kubo formulae.
The calculated transport coefficients with only the update of atomic charges remain less accurate. The accuracy is also imbalanced between viscosity and electrical conductivity. In contrast, the fully-optimized force field including van der Waals parameters provides good agreements for them with the experimental data, and moreover, the obtained accuracy is consistent over the various ionic liquids. Therefore, it turns out that the suggested optimization scheme of non-polarizable force field is useful to predict transport properties of various ionic liquids. In this oral presentation, the detail strategy of force-field development and the calculated structural/transport properties will be explained.
[1] CP2K, version 4.1, The CP2K Foundation, Zürich, 2014.
[2] P. E. Blöchl, J. Chem. Phys. 103, 7422 (1995).
[3] N. C. Lopes, J. Deschamps, A. H. Padua, J. Phys. Chem. B 108, 2038 (2004).
Ionic liquids are salts with melting points below ambient temperatures. They are nonvolatile and nonflammable and solve various chemicals by chemical modifications. Ionic liquids have attracted much attention as potential gas absorption media. In order to use ionic liquids for the chemical processes involved with gas absorption, the understanding of the gas absorption property and mechanism is of primarily importance. This talk presents some of highlight data on gas absorption in the ionic liquids.
CO2 absorption property and mechanism were investigated for ionic liquids based on the cations 1-ethyl-3-methylimidazolium ([emim]+), N,N-diethyl-N-methyl-N-heptyl-ammonium ([N1227]+), N,N-diethyl-N-methyl-N-(6-hydroxy)hexyl-ammonium ([N122,6OH]+), and dodecyltributylphosphonium ([P444,12]+) and the acetate anion ([AcO]-). Each ionic liquid absorbed CO2 chemically and desorbed it reversibly. The largest CO2 solubility and temperature dependency at 313.2 K were observed in [emim][AcO] and [N122,6OH][AcO], respectively. The 13C NMR spectra revealed that CO2 was absorbed in ionic liquids with different absorption mechanisms, namely, the CO2-carbene ([emim][AcO]), CO2-alkoxide ([N122,6OH][AcO]), CO2-acetate ([N1227][AcO]), and CO2-phosphineylide ([P444,12][AcO]) complexes.
We measured high-pressure CO2 solubilities in [emim]+ ionic liquids with [AcO]-, 2-methoxyethoxyacetate ([C1OC2OC1CO2]-), and 3-(2-methoxyethoxy)propionate ([C1(OC2)2CO2]-) to investigate the effect of ether modification on the CO2 physical and chemical absorption. We success fully deconvoluted the high-pressure CO2 solubility into the contributions of physical and chemical absorption. The validity of this numerical analysis was quantitatively confirmed by the NMR spectra for the [emim][[AcO]+CO2 mixtures at high pressures, which can separately identify the chemical and physical species. According to the deconvolution results, the 2-methoxyethoxy group improved the physisorption (in mole fraction) and worsened the chemisorption compared to the unmodified acetate, whereas the negative effects on both the absorptions (in mole fraction) were observed by the 2-methoxyethoxymethyl modification.
I will talk also about the NH3 and water vapor absorption in the ionic liquids in the presentation.
